

1 Introduction
We have recently reported that the trinuclear Fe–Hg–Fe chain complex 1 is a very versatile building block in heterometallic chemistry because of the presence of two phosphine groups available for further complexation to a metal centre and of the possible rotation of the iron fragments about the metal–metal σ bonds [1]. This confers considerable flexibility to this diphosphine-type metalloligand. We were thus able to show that in the presence of the Cu(I) complex [Cu(NCMe)4]PF6, an equilibrium exists in solution between an open chain system 2 and the cyclic structure 3, which can be shifted to either side as a function of the concentration in Cu(I) complex (Fig. 1).
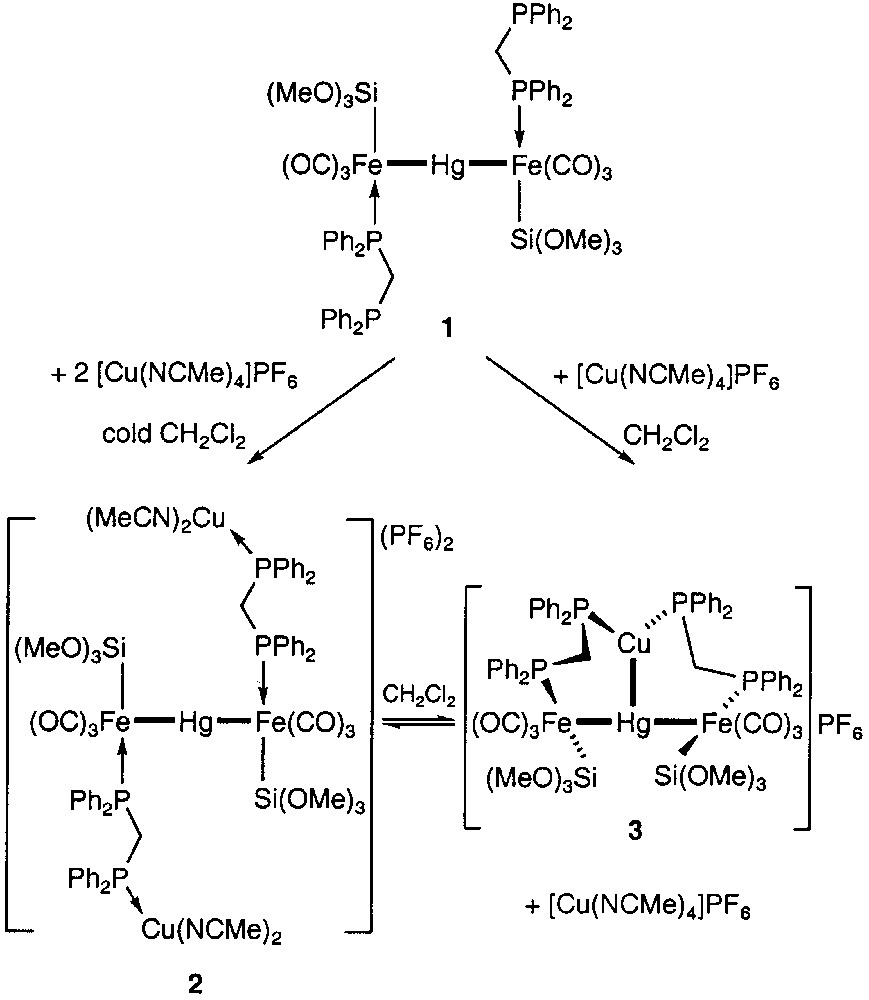
Reactions of the trinuclear metalloligand 1 with [Cu(NCMe)4]+.
The structural characterization of complex 3 revealed the unexpected presence of a Cu–Hg interaction [2.689(2) Å] between the two chemically different d10 closed-shell ions and established that the conformation of polymetallic mesocycles may be controlled by metal–metal bonding. Furthermore, the rare dynamic behaviour of this complex in solution was compared to that of a ‘molecular torsion pendulum’ [1].
2 Results and discussion
We have now examined whether Hg(II), another d10 closed-shell ion, could also be coordinated by the phosphorus lone pairs of 1 and afford a 10-membered ring structure, like the isoelectronic Cu(I) ion. Reaction of 1 with HgCl2 in the presence of 2 equiv TlPF6 in THF did not lead to the expected analogue of 3 with Hg(II) in place of Cu(I). Spectroscopic data indicated instead the formation of the known dinuclear complex 4 [2] (Fig. 2). This could be explained by a transmetalation reaction with migration of one iron metalate fragment from 1 to the Hg(II) centre. Obviously, chloride redistribution between the two Hg centres occurred in preference to abstraction by TlPF6.
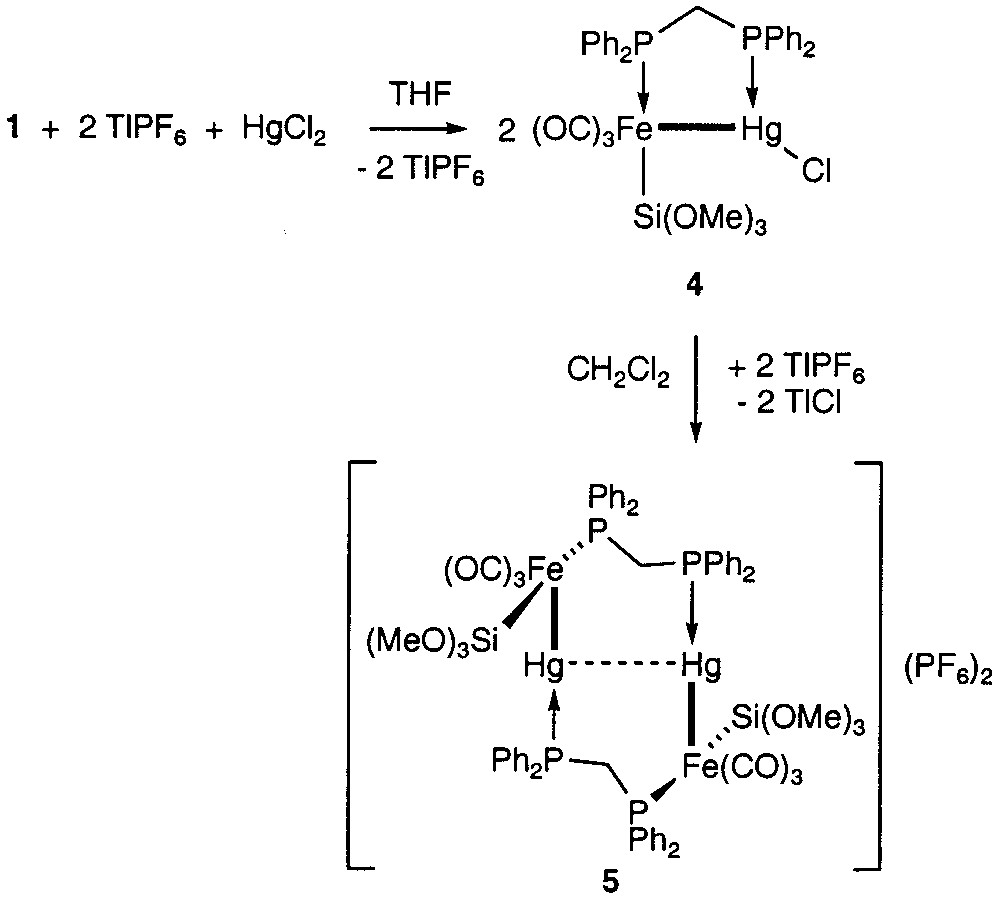
Reactions of 1 with Hg2Cl2.
Related transmetalation reactions have been observed before and are known to be rather frequent in mercury chemistry [3]. The broadening of the 31P NMR signal of the mercury bound phosphorus in 4 suggests possible rapid coordination/decoordination equilibrium of the chloride abstractor Tl+ to the Hg-bound Cl. However, evaporation of the THF reaction mixture to dryness and dissolution of the solid residue in CH2Cl2 afforded a yellow solution and a white precipitate of TlCl. A new, dicationic complex 5 was identified by 31P NMR spectroscopy and assigned the structure shown in Fig. 2. It formally results from dimerisation of the cation generated from 4, which leads to a tetranuclear metal core. A pronounced downfield shift of the signal for the Hg-bound phosphorus accompanied by a remarkably small 1J (Hg–P) coupling constant is indicative for a trans P–Hg–M arrangement (M = transition metal) in 5 [4], the [AX]2 pattern of the entire 31P NMR spectrum accounts for the dimeric structure. A similar structure has been found in the isoelectronic complex [AuFe{Si(OMe)3}(CO)3(μ-dppm)]2, which was shown by X-ray crystallography to contain a d10–d10 interaction between the gold centres [5]. Since the new complex 5 decomposed within hours in solution, we have so far not been able to grow crystals to establish whether or not a d10–d10 interaction exists between the mercury centres. On the basis of the 31P NMR data (Fig. 3), some Hg–Hg d10–d10 interactions may be suggested, because the Hg-bound phosphorus shows two sets of mercury couplings and the coupling constant J(PHg, PHg′) is positive.

Experimental (lower trace) and simulated (upper trace) 31P NMR spectrum of 5(OTf)2. Right part: PHg; left part: PFe.
The same complex 5 (as its OTf derivative) was formed when 1 was reacted with Hg(OTf)2, itself prepared from HgCl2 and 2 equiv AgOTf. This raises the suggestion that an Hg-relative to 3 may be formed in situ, which is thermodynamically unstable, and rearranges to give 5 by transmetalation and phosphine migration (Fig. 4).

Possible pathway to the formation of 5.
It was found that complex 1 possesses approximate C2 symmetry, the pseudo-C2 axis being perpendicular to the trimetallic chain and passing through the mercury atom [5]. The two uncoordinated phosphine moieties are very far away from the Hg centre, although the formation of (weak) interaction between a phosphine donor and Hg would have led to a favourable five-membered ring. To examine the possibility for intramolecular coordination of a functional end of the dangling ligand, we have now prepared the disulphide derivative of 1 (Fig. 5), which appeared to be a good candidate to develop a dative interaction between the sulphur and the soft mercury ion in a six-membered ring. The reaction of 1 with elemental sulphur in THF led to 6 in 94% yield.
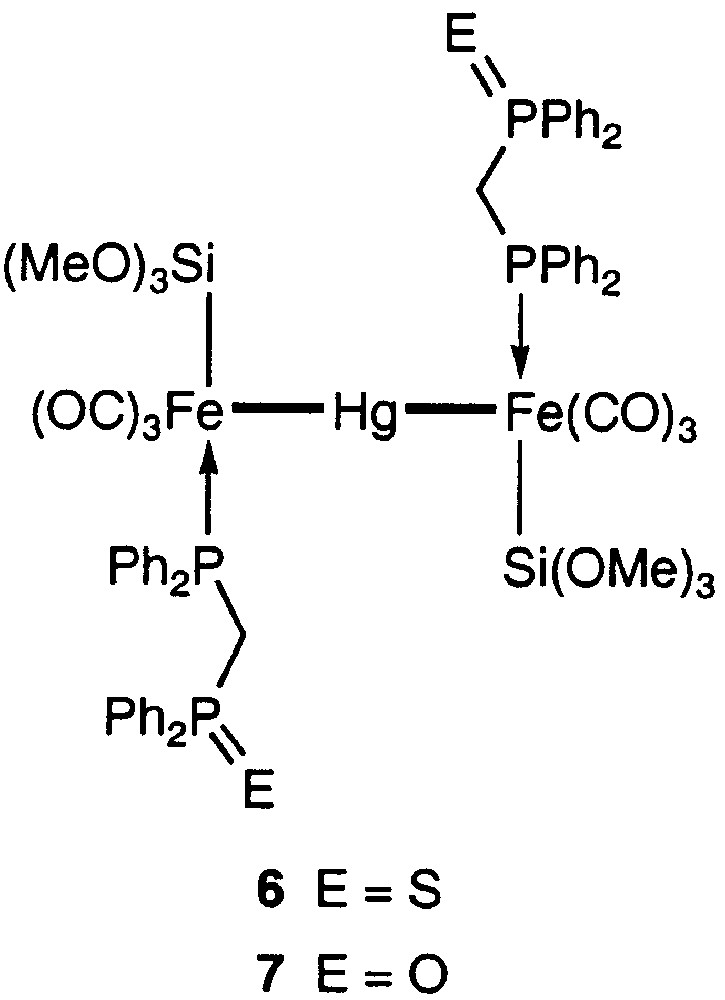
Products of the reaction between 1 and S or H2O2, respectively.
A single crystal X-ray diffraction study of 6·CHCl3was performed (Fig. 6) and selected bond distances and angles are given in Table 1. There is no S–Hg contact in the solid state, neither intra- nor intermolecular. This does not mean of course that such interactions cannot develop in solution. A variable-temperature 31P{1H} NMR study revealed in fact that there is an equilibrium between two different structures in solution: at room temperature, two broad resonances for the Fe-bound and S-bound phosphorus atoms, respectively, indicate a dynamic behaviour for compound 6. At 253 K, however, two sets of signals with relative intensities 95/5 are observed. The major compound (6a) displays an [AX]2 pattern that is assigned to the open-chain structure found in the solid state. The minor isomer (6b) exhibits an AMPX spin system and this is attributed to the formation of a six-membered ring resulting from coordination of one dppm-S ligand to mercury via its sulphur donor site (Fig. 7).
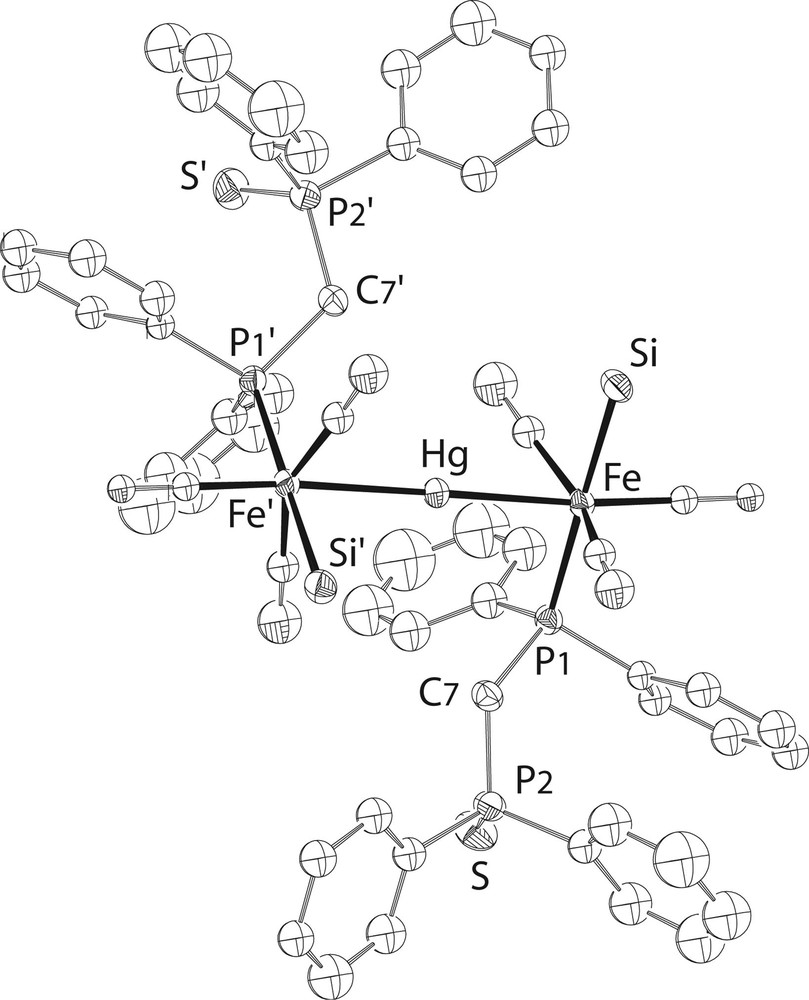
View of the structure of the trinuclear complex in 6•CHCl3. The OMe groups at Si have been omitted for clarity.
| 6•CHCl3 | 7 | |
| Fe–Hg | 2.581(1) | 2.6195(5) |
| Fe–P(1) | 2.268(1) | 2.268(1) |
| Fe–Si | 2.308(1) | 2.311(1) |
| P(2)–S/O | 1.941(2) | 1.551(4) |
| Fe–Hg–Fe | 175.76(3) | 179.05(2) |
| P(1)–Fe–Si | 178.88(4) | 176.16(5) |
| P(1)–C(7)–P(2)–S/O | 47.5(2) | 29.0(3) |
| C(7)–P(1)–Fe–Hg | 41.5(1) | 173.3(1) |

Dynamic behaviour of complex 6 in solution.
By progressively raising the temperature, a pronounced shift of the weighted average 31P NMR signals to the mean value of the respective resonances attributed to 6b was observed. This indicates a thermodynamic preference for the ring structure 6b over the open-chain structure 6a at elevated temperatures.
For comparison, we also prepared the bis(phosphine monoxide) analogue, 7 (Fig. 5) and neither the NMR study nor the crystal structure determination indicated any oxygen–mercury interaction (see Fig. 8 andTable 1 ). This is actually less surprising than in the case of the bis(phosphine sulphide) complex. The closest contact between the phosphine-oxide oxygen atom and a hydrogen atom is an intermolecular contact of 2.384 Å involving H(27), situated on a phenyl ring. It is interesting that in comparison with 6, one observes in 1 [5] and in both dppm-O ligands of 7 a twist by ca 130° about the Fe–P bond, resulting in an elongation of the molecule in the directions of the Fe–Hg–Fe axis. This may best be seen by comparison of the C(7)–P(1)–Fe–Hg torsion angles, which amount to 41.53(11)° (6) and 173.30(13) (7), respectively.
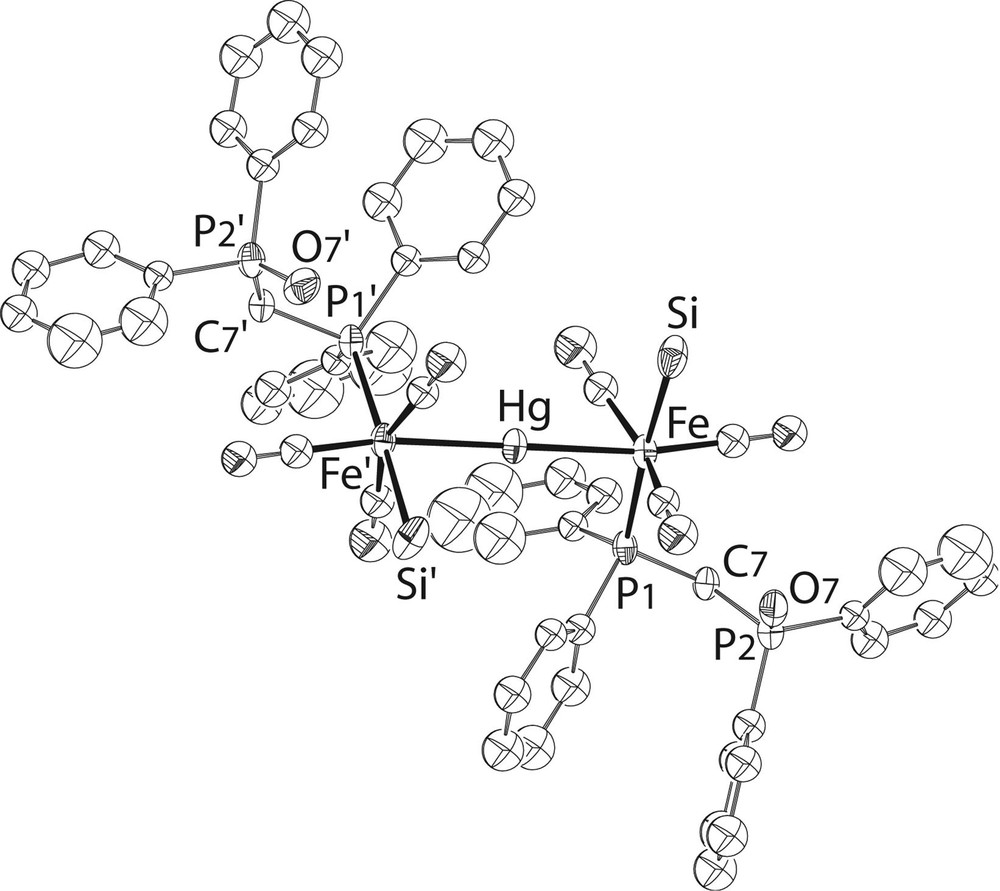
View of the structure of 7. The OMe groups at Si have been omitted for clarity.
When compound 6 was reacted with complexes of the univalent cations Cu(I), Ag(I) or Au(I), in all three cases insoluble precipitates were obtained and precipitation was accompanied by the formation of metallic powders. The products appear to be thermally labile at room temperature, which is most pronounced in the cases of Ag and Au. Their insolubility suggests the formation of coordination polymers.
3 Experimental
3.1 General
If not otherwise stated, reactions were carried out under nitrogen, in dried and distilled solvents. Compound 1, dppm, [Cu(NCMe)4]BF4 and [Au(THT)Cl] were prepared according to published procedures [5,6–8], all other starting materials were purchased from commercial suppliers and used without further purification. The 1H and 31P NMR spectra were recorded on Bruker AV300 and AM300 instruments, chemical shifts are reported in ppm and were referenced to TMS (1H) and external 85% H3PO4 (31P), respectively, coupling constants are reported in Hertz. IR spectra were recorded on a Perkin Elmer FT-IR 1600 instrument.
3.2 Synthesis of the complexes 5, 6 and 7
3.2.1 Compound 5(PF6)2
To a solution of 1 (192 mg, 0.129 mmol) and TlPF6 (104 mg, 0.3 mmol) in 20 ml of THF was added solid HgCl2 (35 mg, 0.129 mmol) with vigorous stirring and a pale yellow solution of 4 formed immediately. After evaporation to dryness, the solid residue was dissolved in CH2Cl2 (5 ml) resulting in a yellow solution of 5 and precipitation of TlCl. The solution was separated from the precipitate by canula filtration. Compound 5(PF6)2 rapidly decomposes in solution by extrusion of elemental mercury and could not be obtained analytically pure in the solid state.
31P{1H} NMR (CD2Cl2): δ = 79.2 (br, 1J(Hg,P) = 860 Hz, PHg), 43.5 (br, 2J(Hg,P) = 280 Hz, PFe), –144.1 (sept., 1J(P,F) = 713 Hz, PF6).
3.2.2 Compound 5(OTf)2
To solid 1 (100 mg, 0.067 mmol) was added a mixture of HgCl2 (18 mg, 0.067 mmol) and AgOTf (34 mg, 0.135 mmol) in THF/CH2Cl2 1:1 (3 ml) with vigorous stirring. The resulting suspension was evaporated to dryness and CH2Cl2 (2 ml) was added. The solution was separated from the precipitate (AgCl) by canula filtration. Compound 5(OTf)2 rapidly decomposes in solution by extrusion of elemental mercury and could not be obtained analytically pure in the solid state.
31P{1H} NMR (CD2Cl2): δ = 77.7 (2J(PHg,PFe) = 26.2 Hz, 3J(PHg,PFe′) = –16.8 Hz, 3J(PHg,PHg′) = 4.6 Hz, 1J(Hg,PHg) = 548 Hz, 2J(Hg,PHg′) = 80 Hz,PHg), 43.5 (4J(PFe,PFe′) = 0, 2J(Hg,PFe) = 303 Hz, 3J(Hg,PFe′) = 0, PFe). Spectral simulation was performed by WinDaisy [9](4J(PFe,PFe′) and 3J(Hg,PFe′) were kept at 0 Hz, linewidth were fixed to 6 Hz and 25 Hz for the isotopomers without and with one active 199Hg, respectively; calculated standard deviations for chemical shifts and P–P coupling constants < 0.1 Hz, for Hg–P coupling constants < 1 Hz).
3.2.3 Compound 6
To a solution of 1 (1.885 g, 1.26 mmol) in 50 ml of THF was added elemental sulphur (81 mg, 2.52 mmol) with vigorous stirring. After 4 h, the resulting suspension was evaporated to dryness, the solid residue was dissolved in CH2Cl2 (100 ml) and filtered through Celite. The filtrate was concentrated to 20 ml and 6 was precipitated by addition of 200 ml of pentane. After filtration the solid product was dried in vacuo. Yield: 1.84 g (94%). Crystals of 6·CHCl3 were grown from CHCl3/pentane. Anal. calc. for C62H62Fe2HgO12P4S2Si2·CHCl3 (M = 1675.0): C, 45.18; H, 3.79. Found: C, 45.65; H, 3.89%.
IR (CHCl3): ν (cm–1) = 2018(w), 1996(s), 1946(sh), 1932(s, br). 1H NMR (CDCl3): δ = 7.6–7.1 (m, 40H, Ph), 3.81 (dd, 2J(P,H) = 10.6, 5.3 Hz; 4H, CH2), 3.55 (s, 18H, (MeO)3Si). 31P{1H} NMR (CD2Cl2, 253 K): δ = 47.5 (PFe), 34.5 (PS), [AX]2 spin system, N ≈ 2J(PFe,PS) = 25 Hz, major compound, ca 95%; δ = 55.3 (d, 2J(P,P) = 30 Hz, PFe), 44.3 (s, br, PFe′), 33.6 (d, 2J(P,P) = 30 Hz, PS), 33.0 (s, br, PS′), minor compound, ca 5%. 31P{1H} NMR (CD2Cl2, 298 K): δ = 49.1, 33.5 ([AX]2 spin system, br, N = 21 Hz). 31P{1H} NMR (CDCl3, 318 K): δ = 50.7, 33.8 ([AX]2 spin system, N = 21 Hz).
3.2.4 Compound 7
To a solution of 1 (192 mg, 0.129 mmol) in CH2Cl2 (10 ml) was added water (0.5 ml) and three drops of H2O2 (35% solution in H2O) under vigorous stirring. After 1 h, the reaction mixture was quenched with water (3 × 2 ml) and the resulting CH2Cl2 solution was filtered through cotton wool. After concentration of the solvent to 1 ml, compound 7 was precipitated by addition of pentane (10 ml), collected by filtration and dried in vacuo. The crude product was recrystallised from CH2Cl2/pentane by vapour diffusion. Yield: 58 mg (29%). Anal. calc. for C62H62Fe2HgO14P4Si2 (M = 1523.5): C, 48.88; H, 4.10. Found: C, 49.23; H, 4.22%.
31P{1H} NMR (CD2Cl2, 298 K): δ = 47.0 (PFe), 23.2 (PO), [AX]2 spin system, N ≈ 2J(PFe,PO) = 13 Hz.
3.3 Reactions of 6 with complexes of Cu(I), Ag(I) and Au(I)
To a solution of 6 (84 mg, 0.054 mmol) in 5 ml of CH2Cl2 was added solid [Cu(NCMe)4]BF4 (17 mg, 0.054 mmol) with vigorous stirring. After 5 min, a brown precipitate was formed, which was insoluble in all common organic solvents.
To a solution of 6 (156 mg, 0.1 mmol) in 5 ml of CH2Cl2 was added solid AgOTf (26 mg, 0.1 mmol) with vigorous stirring at 0 °C. A brown precipitate immediately formed, which was insoluble in all common organic solvents. The precipitate becomes grey upon standing, which was attributed to formation of metallic silver.
To a solution of 6 (156 mg, 0.1 mmol) in 5 ml of CH2Cl2 was added solid [Au(THT)Cl] (32 mg, 0.1 mmol) with vigorous stirring at 0 °C. A brown precipitate immediately formed, which was insoluble in all common organic solvents. The precipitate rapidly becomes black upon standing, which was attributed to formation of metallic gold.
3.4 X-ray data collection
Crystals of 6·CHCl3 and 7 suitable for X-ray diffraction were grown from CHCl3/pentane or CH2Cl2/pentane, respectively. Crystals were mounted on a Kappa CCD diffractometer, data were collected using phi-scans and the structure was solved by direct methods and refined against F2 using SHELX 97 software [10,11]. A total of 16 286 (6·CHCl3)/9157 (7) reflections were collected, of which 12 839 (6·CHCl3)/6624 (7) unique reflections had intensities I > 2 σ(I). All non-hydrogen atoms were refined anisotropically, except for the solvate molecule in 6·CHCl3, H atoms were placed at calculated ideal positions. Final R-values were R = 0.0398, wR2(all data) = 0.1084 (for 6·CHCl3) and R = 0.0438, wR2 (all data) = 0.1172 (for 7), respectively. Full data collection parameters, and structural data are available as supplementary material.
4 Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as cif file Nos CCDC 188953 (7) and 188954 (6·CHCl3), and can be obtained by contacting the CCDC.
Acknowledgements
This work was supported by the CNRS and the French ‘Ministère de la Recherche’. W.S. is grateful to the ‘Fonds zur Förderung der wissenschaftlichen Forschung’ (FWF, Austria) for a post-doctoral grant (Erwin-Schrödinger-Stipendium Proj. No. J2021) and to Prof. Paul Peringer for access to WinDaisy facilities.