

1 Introduction
The search for molecule-based materials exhibiting multiphysical properties is one of the current challenges in molecular materials science [1, 2]. Great interest is currently devoted to obtain organic/inorganic hybrid materials in which it is expected that there is long-range magnetic coupling between the localised spins on d orbitals of the paramagnetic transition metal ions of the inorganic part through the mobile electrons of the conducting networks (π-electrons) [3]. Consequently, our strategy involves the covalent attachment of metal-ion binding groups to BEDT–TTF derivatives and their linkage into supramolecular systems. With the appropriate choice of metal ions, the resulting system would be expected to show multiple physical properties, such as electrical conductivity or superconductivity and magnetic effects, optical and magnetic properties or spin crossover, in a synergistic way. Based on the chemistry of thiapendione, synthetic approaches to symmetrically functionalised BEDT–TTF derivatives have been realised in our group in order to prepare novel organic conducting materials featuring TTF moieties within metal binding ligand systems [4]. Herein, we describe a short and efficient synthetic route to two new unsymmetrically functionalized BEDT–TTF derivatives 4 and 5, together with their electrochemical properties as well as the crystal structure of compound 4.
2 Results and discussion
2.1 Syntheses
In general, several strategies for the preparation of unsymmetrically functionalised BEDT–TTF derivatives are possible [5]. The most widely used method is via the phosphite-mediated cross coupling of two appropriate chalcogenones leading to a mixture from which the desired species must be separated from the symmetrical by-products [6]. However, some BEDT–TTF derivatives are not successfully available by the simple procedure of refluxing their corresponding thiones/ones in the presence of a phosphorous (III) compound, [P(OMe)3, P(OEt)3 or P(Ph)3] [7]. Furthermore, this classical synthesis is problematic, suffering from disadvantages, such as low yields, tedious separations, multistep procedures, and/or expensive starting materials [8]. Therefore, an alternative strategy has been adopted by several groups. The key compound in this case is the 4,5-bis(2′-cyano-ethylsulfanyl)-4′,5′-ethylenedithiotetrathiafulvalene 3, which can be readily prepared as described [9] by treatment of equimolecular amounts of 4,5-ethylenedithio-1,3-dithiole-2-thione 1 and 4,5-bis(2′-cyanoethylsulfanyl)-1,3-dithiol-2-one 2 at 100 °C in neat P(OEt)3 (Fig. 1). Then compound 3 can simply be deprotected by a basic reagent [10] and, subsequently, by appropriate alkylations, to give two new functionalized BEDT–TTF derivatives 4 and 5 in good yields (Fig. 2).
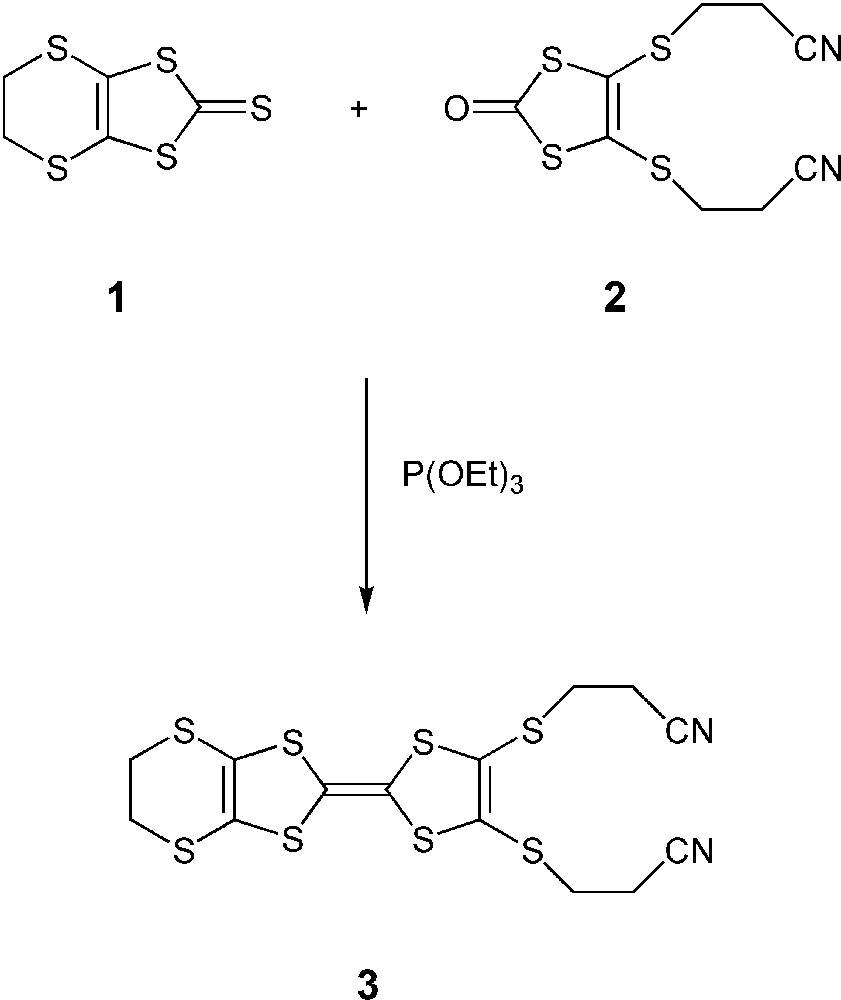
A synthetic route for the preparation of key compound 3.

A short and efficient synthetic route for the preparation of BEDT–TTF derivatives 4 and 5.
2.2 Electrochemical studies
The redox potentials of new donors 4 and 5 were measured in dichloromethane by cyclic voltammetry. Their cyclic voltammetric data are collected in Table 1 together with those of BEDT–TTF for comparison. In each case, two reversible single-electron oxidation waves are observed, typical of the TTF system corresponding to E1/21 and E1/22 in Table 1. In general, the values of 4 and 5 are slightly raised as compared to those of BEDT–TTF, which is probably due to the electron-withdrawing effect of the pyridine and pyrazine units. These results also show that the presence of the pyridine or pyrazine units relatively far away from the TTF core on the side chains –SCH2–C5H4N and –SCH2–C4H3N2 respectively, has only a very weak influence on their values.
Cyclic voltammetric dataa
| E (V) | 4 | 5 | BEDT–TTF |
| E1/2 1 | 0.58 | 0.56 | 0.52 |
| E1/2 2 | 0.97 | 0.90 | 0.94 |
2.3 Crystal structure of donor 4
BEDT–TTF derivatives 4 and 5 have been characterized by elemental analysis, 1H NMR and 13C NMR as well as IR spectroscopy (see the Experimental section). The single crystal X-ray structure of 4 is depicted in Fig. 3 and the selected bond distances and angles are given in Table 2. TTF core of compound 4 is nearly planar with a dihedral angle of 1.94(9)° between the two five-membered rings (S1, S2, C1, C7, C8 and S3, S4, C2, C3, C6).

ORTEP drawing of compound 4 showing 50% probability thermal ellipsoids and the atom-numbering scheme.
Selected bond distances (Å) and bond angle (°) for 4
| S(1)–C(1) | 1.7508(16) | S(6)–C(5) | 1.805(2) |
| S(1)–C(8) | 1.7511(16) | S(7)–C(7) | 1.7454(16) |
| S(2)–C(7) | 1.7544(16) | S(7)–C(9) | 1.824(2) |
| S(2)–C(1) | 1.7552(16) | S(8)–C(8) | 1.7490(16) |
| S(3)–C(6) | 1.7501(17) | S(8)–C(15) | 1.8297(19) |
| S(3)–C(2) | 1.7559(16) | C(1)–C(2) | 1.341(2) |
| S(4)–C(3) | 1.7489(16) | C(3)–C(6) | 1.340(2) |
| S(4)–C(2) | 1.7540(16) | C(4)–C(5) | 1.507(3) |
| S(5)–C(3) | 1.7457(17) | C(7)–C(8) | 1.345(2) |
| S(5)–C(4) | 1.7991(18) | C(9)–C(10) | 1.494(3) |
| S(6)–C(6) | 1.7450(16) | C(15)–C(16) | 1.498(2) |
| C(1)–S(1)–C(8) | 95.71(7) | C(7)–S(7)–C(9) | 100.20(8) |
| C(7)–S(2)–C(1) | 95.59(8) | C(8)–S(8)–C(15) | 101.55(8) |
| C(6)–S(3)–C(2) | 94.80(8) | C(2)–C(1)–S(1) | 122.98(13) |
| C(3)–S(4)–C(2) | 95.09(8) | C(2)–C(1)–S(2) | 122.99(13) |
| C(3)–S(5)–C(4) | 99.86(8) | S(1)–C(1)–S(2) | 113.98(9) |
| C(6)–S(6)–C(5) | 101.42(8) | C(1)–C(2)–S(4) | 122.61(13) |
| C(1)–C(2)–S(3) | 122.43(13) | S(4)–C(2)–S(3) | 114.92(9) |
In the crystal lattice, the molecules are linked by a network of some unconventional C–H···N and C–H···S hydrogen bonds (Fig. 4 and Table 3). Interestingly, the ‘BEDT–TTF’ and the pyridine moieties of 4 are arranged in two separate columns. Within the ‘BEDT–TTF column’, some S···S contacts in the range 3.832–4.296 Å are observed, and the interplanar separation of 6.03 Å is rather large, dictated by the bulky substituents (Fig. 5). In the ‘pyridine column’, the pyridine rings involving atom N2, of symmetry related molecules, are separated by an inter-planar stacking distance of ca 4.02 Å.
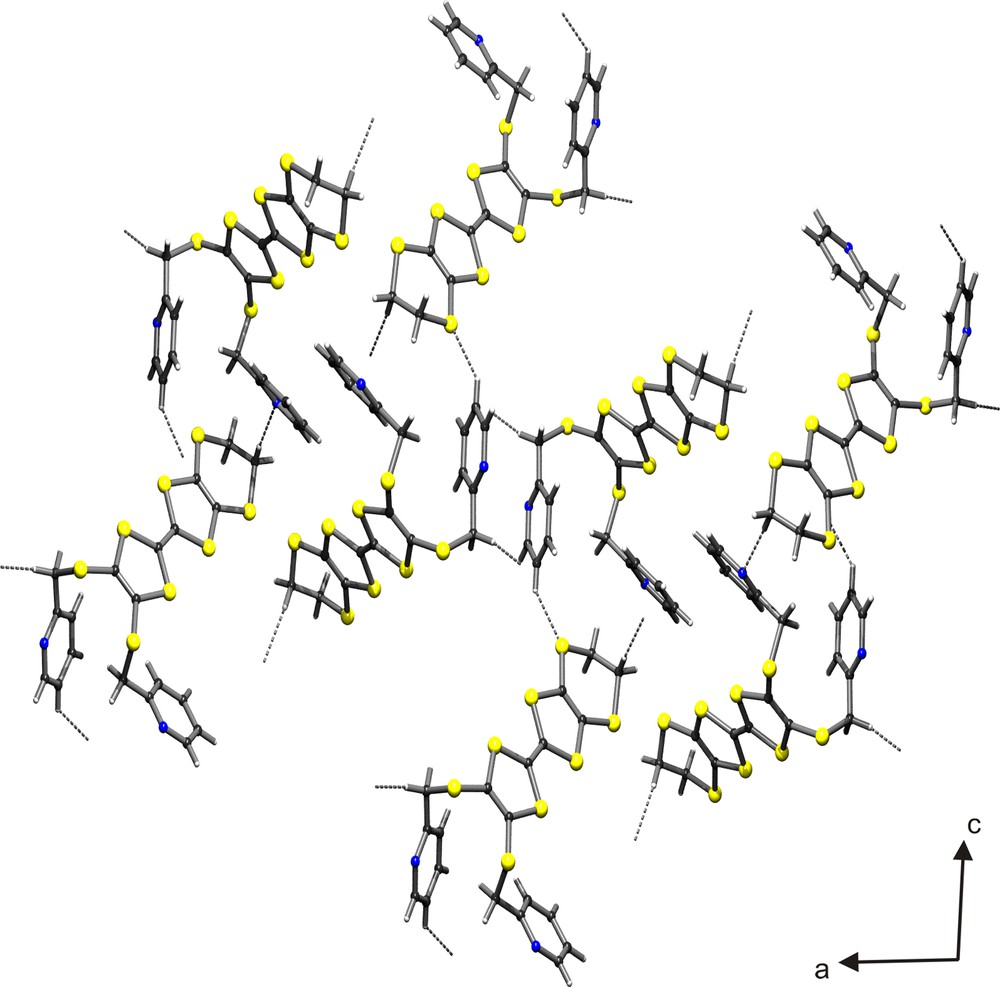
Crystal structure (ac projection) of compound 4 showing the unconventional hydrogen bonds.
Some unconventional hydrogen bond distances [Å] and bond angles [°] for 4
| Donor–H···Acceptor | D···A | D–H···A |
| C(4)–H(4B)···N(2) | 3.487(2) | 170.45 |
| C(9)–H(9B)···N(1) | 3.339(3) | 147.50 |
| C(13)–H(13A)···S(6) | 3.550(2) | 133.53 |

Crystal packing (ac projection) of compound 4.
In conclusion, two novel unsymmetrically functionalized BEDT–TTF derivatives 4 and 5 were synthesized and characterized. Their redox potentials were determined by cyclic voltammetry in CH2Cl2 solution and results revealed they were slightly weaker donors than BEDT–TTF. A crystal structure analysis was carried out for compound 4, showing an interesting molecular packing pattern. The binding ability to transition metal ions, the formation of charge-transfer complexes and ion radical salts of these promising new donors is currently under investigation in our laboratory and will be published in due course.
3 Experimental Section
3.1 Synthesis
3.1.1 General methods and instrumentation
Reactions requiring air- and/or water-sensitive manipulations were conducted under argon with dry, freshly distilled solvents. Unless stated otherwise, all other reagents were purchased from commercial sources and used without additional purification. 2-chloromethylpyrazine [11], 4,5-ethylenedithio-1,3-dithiole-2-thione (1) [12], 4,5-bis(2′-cyanoethylsulfanyl)-1,3-dithiol-2-one (2) [12] and 4,5-bis(2′-cyano-ethylsulfanyl)-4′,5′-ethylenedithiotetrathiafulvalene (3) [9] were prepared according to literature procedures. Unless otherwise specified, all 1H and 13C NMR spectra were measured in CDCl3 at 300 MHz and 75 MHz, respectively. Melting points are reported in degrees Celsius and are uncorrected. Elemental analyses were performed on an EA 1110 Elemental Analyser CHNS Carlo Erba Instruments. FT–IR data were collected at a resolution of 4 cm–1. Cyclic voltammetric measurements were conducted on a VA-Stand 663 electrochemical analyser. Mass spectra were recorded on a Micromass AutoSpec spectrometer using FAB or EI. Analytical TLC was carried out on Merck Silica gel 60 F254 coated aluminium foil; the same type of silica was used for columns.
3.1.2 Thiolate deprotection-alkylation procedure
A solution of 0.5 M EtONa/EtOH (8 ml) was added with a syringe to a suspension of 4,5-bis(2′-cyano-ethylsulfanyl)-4′,5′-ethylenedithiotetrathiafulvalene (3) (0.465 g, 1 mmol) in anhydrous degassed EtOH (27 ml) under Argon. After being stirred at room temperature for 4 h, the mixture was reacted with a solution of the appropriate alkyl halide (12 mmol) in anhydrous degassed EtOH (24 ml), and then the mixture was stirred overnight. H2O was added to quench the reaction, and the reaction mixture was then extracted with CH2Cl2. The combined organic extracts were washed, dried over MgSO4, filtered, and concentrated in vacuo to give a dark-brown oil which was purified by flash chromatography on silica gel, initially with CH2Cl2 (to remove any unreacted alkyl halide as well as unwanted by-products), and then with 10:1 CH2Cl2/ethyl acetate, unless mentioned, to give the desired product.
4,5-bis(2-pyridylmethylsulfanyl)-4′,5′-ethylenedithiotetrathiafulvalene (4). Yield: 80%. Colour: brown. Melting point: 105 °C. 1H NMR: δ 3.29 (s, 4H), 4.02 (s, 4H), 7.14–7.25 (m, 4H), 7.59–7.65 (dt, J = 2 Hz, J = 8 Hz, 2H), 8.54–8.55 (d, J = 4 Hz, 2H); 13C NMR: 156.4, 149.7, 136.7, 129.2, 123.3, 122.4, 113.9, 113.6, 108.7, 42.3, 30.2. IR (KBr, cm–1): 1590, 1568, 1471, 1435, 1406, 1149, 1085, 1046, 994, 771, 743. Mass spectrum (EI) m/z: 540 (M+). Anal. calcd for C20H16N2S8: C, 44.42; H, 2.98; N, 5.18. Found: C, 44.71; H, 2.96; N, 5.18.
4,5-bis(2-pyrazylmethylsulfanyl)-4′,5′-ethylenedithiotetrathiafulvalene (5). After work-up, the resulting dark-brown residue was subjected to column chromatography, eluting initially with a gradient of 20–100% ethyl acetate in CH2Cl2 and then with MeOH / ethyl acetate (1:2) to give 5. Yield: 63%. Colour: Dark brown. Melting point: 90 °C. 1H NMR: δ 3.29 (s, 4H), 4.03 (s, 4H), 8.50 (m, 6H); 13C NMR: 152.4, 144.6, 144.3, 143.4, 129.2, 113.8, 111.9, 110.5, 39.4, 30.1; IR (KBr, cm–1): 1526, 1473, 1400, 1287, 1215, 1173, 1112, 1055,1018, 881, 772. Mass spectrum (FAB) m/z: 542 (M+). Anal. calcd for C18H14N4S8·0.25EtOAc: C, 40.40; H, 2.85; N, 9.91. Found: C, 40.50; H, 2.73; N, 9.85.
3.2 X-ray crystallography
C20H16N2S8·0.5 (C4H8O2), MW = 584.88, monoclinic, space group C2/c, a = 36.0971(19), b = 6.0257(2), c = 22.8424(13) Å, β = 92.850(4)°, V = 4962.3(4) Å3, Z = 8, dcalc = 1.566 g cm–3, μ(Mo Kα) = 0.740 mm–1. 28 000 reflections measured, 5447 independent reflections, 4744 observed reflections (I > 2 σ(I)), final R1 = 0.0303 (observed data), wR2 = 0.827 (all data).
Suitable crystals were obtained as red-brown plates by slow evaporation of a CH2Cl2 / CH3COOEt solution of 4. The intensity data were collected at 153 K (–120 °C) on a Stoe Mark II–Image Plate Diffraction System [13] using Mo Kα graphite monochromated radiation (λ = 0.710 73 Å). Image plate distance 120 mm, ω oscillation scans 0–180° at φ 0°, and ω oscillation scans 0–60° at φ 90°, step Δω = 1°, 2θ range 1.91–54.78°, dmax – dmin = 21.331 – 0.772 Å. The structure was solved by direct methods using the programme SHELXS-97 [14]. The refinement and all further calculations were carried out using SHELXL-97 [15]. The hydrogen atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least squares on F2. A series of electron density peaks were located along the 2-fold axes, parallel to the crystallographic b axis, but it could not be identified clearly as a particular solvent molecule, for example ethylacetate. The SQUEEZE routine in PLATON [16] was used to modify the HKL file, and indicated an empty volume of approximately 503 Å3 per unit cell, corresponding to 126 electrons per unit cell. This electron density was equated to half a molecule of ethylacetate per molecule of 4.
4 Supplementary material
Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary material No. CCDC 196642 and can be obtained by contacting the CCDC (fax: (+44) 1223 336 033; email: deposit@ccdc.cam.ac.uk).
Acknowledgements
Support for this research by the ESF and TMR Scientific Programme ‘Molecular Magnets’ is gratefully acknowledged.