

1 Introduction
Since the first observation (M. Farady, J. Tyndall) and explanation of the various colours of colloidal gold particles [1], the optical properties of nanosized metal particles continue to attract a great deal of interest [2–6]. During the past years, attention has focused on size-controlled synthesis and self-assembled array of metallic nano-clusters that stems from the application of metal nanoparticles in surface-enhanced spectroscopy, photo-catalysis and wavelength filters [7,8]. A large number of physical and chemical methods, including metal evaporation [9], electro-deposition [10], reduction of metal salts [11] and thermal [12] or sono-chemical [13] decomposition of metal-organic compounds have been developed to produce metal particles in nanometre domain. However, most of these studies deal with noble or transition metals [3] and very little is known about the colloidal nanoparticles of main group elements. We are developing molecular routes for a chemically controlled synthesis of metal and oxide nanoparticles [14–17]. In this paper, we demonstrate the electroless reduction of a lead amide by a well-defined molecular reductant to initiate and control the growth of lead nanoparticles at the molecular scale.
Our approach entails (i) chemically controlled synthesis and homogeneous growth of lead nano-clusters in non-aqueous solutions and (ii) templating the lead clusters either by capping them with organic ligands to obtain stable colloids or by filling the nanoparticles in the cylindrical pores of a porous alumina membrane. The template method using nano-porous membranes is a powerful approach for producing organized nanostructures (e.g., nano-wires or nano-tubes) that display interesting properties due to their reduced dimensionality [18–20]. For instance, superconducting Pb nano-wires can be prepared by electro-deposition in track-etched polymer membranes [21]. We are using porous anodic alumina (PAA) discs as host templates because they are optically transparent in a wide range (0.3–6.0 μm) and the parallel alignment of cylindrical pores allows a defined orientation of metal particles. Both, vacuum-induced infiltration (case a, Scheme 1 ) of the colloidal lead particles as well as the reduction of the lead precursor within the pores of PAA (case b, Scheme 1) were performed in order to obtain the Pb/Al2O3 systems described here.
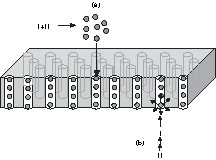
2 Experimental section
2.1 Reagents and instrumentation
The syntheses of lead(II) amide, [Pb{N(SiMe3)2}2] (I) [22], and tert-butoxy alane, [H2Al(OtBu)]2 (II), [23] were done following the established procedures. All experimental manipulations were performed in a modified Schlenk-type apparatus taking stringent precautions against atmospheric moisture. Solvents used were purified by standard methods and stored over appropriate desiccating agents. Room-temperature powder X-ray diffraction patterns were obtained on a STOE diffractometer operating with Cu Kα radiation. The X-ray diffraction on single crystals was performed on an IPDS instrument of STOE, Darmstadt/Germany, using Mo Kα radiation. The infrared spectra were recorded as KBr discs on a BioRad FT-IR-165 spectrometer. The SEM and energy dispersive X-ray (EDX) analysis were carried out in the specimen chamber of an EDX coupled scanning electron microscope CAM SCAN S4. Optical transmission measurements were carried out on a BENTHAM spectrophotometer fitted with a double-monochromator.
2.2 Synthesis of Pb nanoparticles and of {[(Me3Si)2N][tBuO]Al–H}2 (III)
Lead nanoparticles were prepared by a redox reaction between lead(II)bis(trimethylsilyl)amide, [Pb{N(SiMe3)2}2] (I), and the tert-butoxy alane [H2Al(OtBu)]2 (II). In a typical experiment, a THF solution of II (12 mmol l–1) was slowly added dropwise to a cooled (–100 °C) solution of I under continuous stirring. This results in a spontaneous reaction causing the following colour changes: original orange → light yellow → bright yellow → light green → dark green → grey–green. The only exception to the above colour changes was observed when acetonitrile was used as the solvent, which gave a burgundy-red solution of lead particles. The reaction mixture was maintained at –100 °C for 1 h and then left to warm up gradually to room temperature, leading to greyish green suspensions of lead particles. The colloids show a limited stability (< 48 h) at room temperature. Indeed, their decomposition is shown by the precipitation of a grey powder that proved to be metallic lead (evidenced by XRD).
When the reaction was performed at –78 °C, no colloid formation was observed and in all the solvents (tetrahydrofuran, acetonitrile, thiophene, thiophenol, ether, and toluene) used in this study, a fine grey-to-black precipitate was obtained. The precipitate consists of lead nanoparticles that redisperse upon ultrasonic treatments. After removal of the solvent, a non-pyrophoric porous solid was obtained, which was confirmed to be lead by EDX, XRD and chemical analysis. This reaction was reproduced at different concentrations of the starting materials (12, 18, 30 and 60 mmol l–1 of I).
The molecular by-product of the reaction between I and II can be isolated by concentrating the solution (diethyl ether or tetrahydrofuran) after separation from the solid lead-clusters. Using 5 mmol of I and II leads to a precipitation of 1.83 g (70% yield) of {[(Me3Si)2N][tBuO]Al-H}2 (III), which can be purified by recrystallisation from diethyl ether. Analysis performed soon after the reaction shows the presence of another by-product III′ which converts to III within 24 h on standing in diethyl ether. Spectroscopic data of III: 1H NMR (200.13 MHz, toluene) δ, ppm: 0.37 (s, 18 H, Si–C–H), 0.39 (s, 18 H, Si–C–H), 1.42 (s, 18 H, O–C–C–H). 1H NMR (200.13 MHz, diethyl ether) δ, ppm: 0.13 (s, 18 H, Si–C–H), 0.14 (s, 18 H, Si–C–H), 1.45 (s, 18 H, O–C–C–H). 13C NMR (50.32 MHz, toluene) δ, ppm: 5.3 (s, 6 C, Si–
{[(Me3Si)2N][tBuO]Al–H}2 (III) crystallizes in the monoclinic space group P21/c with Z = 2 (a = 11.264(2), b = 14.518(3), c = 10.337(2) Å, β = 99.86(3)°, V = 1665.4(6) Å3). For the record of the diffraction intensities, an IPDS flat scan (STOE) was used with Mo Kα radiation (2.31° < θ > 24.13°). From 10225 reflections, 2566 were unique (Rint = 0.0278) to refine 140 parameters. The structure was solved using direct methods [39] and refined by full-matrix least squares on F2 with anisotropic temperature factors for all non-hydrogen atoms. The final R1-value was 0.0367 (I > 2 σI) and wR2 = 0.1067 with electron density left: 0.291/–0.223 e Å–3 (further details on the crystal structure investigations may be obtained from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ (UK) on quoting the depository number CCDC-232382).
When the reaction between I and II is performed in non-stoichiometric ratios (for example I/II = 2:1) in diethyl ether, the formation of lead particles or colloids was not influenced. Compared to the equimolar reaction of I and II, the solution spectra of the reaction mixtures (1H and 13C NMR) are much more complicated. Indeed, a mixture of compounds could be recognized by their spectral fingerprints, but the compounds could not be separated by simple crystallization. On sublimation of the solid residue, we were able to separate {[(Me3Si)2N][tBuO]Al-H}2 (III), the well-known aluminium amide [(Me3Si)2N]3Al (IV) [40] and the starting material Pb[N(SiMe3)2]2 (I) in an approximate 1:1.5:2 ratio, respectively. The compounds were characterized unambiguously by 1H, 13C and 29Si NMR and by comparison of X-ray data of IV with the published structure [40] (1H NMR (200.13 MHz, diethyl ether) δ, ppm: IV: 0.18 (s, 54 H, Si–C–H). 13C NMR (50.32 MHz, diethyl ether) δ, ppm: IV: 5.89 (s, 18 C, Si–
The molecule {[(Me3Si)2N][tBuO]Al–H}2 (III) can be synthesized by an alternative route: 1.377 g (6.74 mmol) of tert-butoxy alane, II, is mixed with 15 ml (11.62 g, 71.98 mmol) of hexametyldisilazane and heated to 118 °C during 18 h under reflux. The evolution of hydrogen is observed. On cooling, crystals form and grow upon concentration of the solution. Yield of III: 1.32 g (37 %); spectroscopic and other characterizations, see above.
2.3 Infiltration of lead nanoparticles in porous alumina membranes
The alumina membrane used in this study had pores of 60-nm diameter. The pore radius of the alumina membrane was determined by high-resolution electron microscopy. Vacuum-induction technique was attempted to fill the pores of the membrane with lead nanoparticles, but these attempts remained unsuccessful due to the much bigger size of the lead particles. Indeed, after these experiments the diameters of the particles were determined and turned out to be on average ca 200 nm. For this reason, an alternative technique was applied, which consists in the reduction of the Pb(II) precursor within the pores of the membrane. This allows the formation of lead nuclei of controlled-growth via coalescence within the cylindrical pores. However, the distribution was affected by reaction kinetics operative during the diffusion of II into the channels of the membrane.
3 Results and discussion
When the molecular lead(II) amide (I) is reacted under inert atmosphere with a stoichiometric amount of the reducing agent, tert-butoxy alane (II), a chemically-controlled synthesis of lead nanoparticles occurs according to Eq. (1). The mechanism of the reaction has not yet been fully determined, however, the isolation and characterization (NMR, X-ray structure) of the cyclic compound [N(SiMe3)2]2(H)2–Al2(OtBu)2 (III) from the reaction mixture suggests a ligand exchange (hydride vs amide), resulting in the reduction of divalent lead species into the elemental form, apparently through an intermediate lead hydride species.
| (1) |
The driving force of the reaction seems to be the higher enthalpy of Al–N bonds (297 kJ mol–1) compared to those of Pb–N (165 kJ mol–1) [24]. Interestingly, the similar reaction of Me2Si(NtBu)2Sn with the same reducing agent, in which tin clusters are formed, produces the hydride-free Me2Si(NtBu)2Al(OtBu)2Al(NtBu)2SiMe2 as the major product [14]. The different molecular products of the reactions could be due to a higher steric bulk of the hexamethyldisilazyl group compared to cyclic bis(amine) or/and to the connection of the two nitrogen atoms by dimethylsilyl in (tBu)N-SiMe2-N(tBu). To test this hypothesis, the same reaction was run in a molar ratio of 2:1 (I:II) (and even under an excess of Pb[N(SiMe3)2]2) without evidence of a hydride-free aluminium compound. Instead, it was found that scrambling reactions of ligands occur as may be deduced from the isolation of Al[N(SiMe3)2]3 by sublimation of the reaction residue, which consists among others of the excess Pb[N(SiMe3)2]2 (I) and of III. Finally, [tBuOAlH2]2 was reacted with pure hexamethyldisilazane serving as a reaction partner and as a solvent, leading to the formation of {[(Me3Si)2N](H)Al(OtBu)}2 (III) exclusively (see Eq. (2)).
| (2) |
The cyclic compound III, which can be sublimed without decomposition, is of more general interest as there are only, to the best of our knowledge, two examples of tetra-coordinated aluminium bound by H, N, O [25,26]. Most of the amino-oxo-alanes isolated so far have aluminium atoms of coordination numbers 5 and 6 [27–34]. In the 1H, 13C and 29Si NMR solution spectra of III the two trimethylsilyl groups are not equivalent, which underlines the steric crowding within the molecule preventing a free rotation around the Al–N bond. In Fig. 1 , the structure of molecule III is shown, determined by single crystal X-ray diffraction. The centro-symmetric molecule has an Al2O2 ring of almost equal Al–O bond lengths (1.847(1) Å) with tert-butyl groups bonded to the oxygen atoms and hexamethyldisilazyl and hydrogen bonded to the aluminium atoms. The bond lengths and angles are in the expected range [25–34].
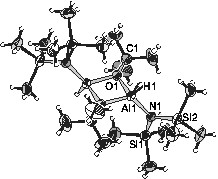
Molecular structure of {[(Me3Si)2N](H)Al(OtBu)}2 (III) with thermal ellipsoids (50%). The non-labelled atoms are either carbon or hydrogen (small balls). Some selected bond lengths (Å) and angles (°): Al1–O1 = 1.847(1), Al1–O1′ = 1.848(1), O1–C1 1.481(2), Al1–H1 = 1.516(6), Al1–N1 = 1.830(2), N1–Si1 = 1.743(2), N1–Si2 = 1.757(2); Al1–O1–Al1′ = 99.41(7), O1–Al1–O1' = 80.59(7), Al1–N1–Si1 = 125.7(1), Al1–N1–Si2 = 114.0(1), Si1–N1–Si2 = 120.0(1), O1–Al1–N1 = 120.21(7), O1'–Al1–N1 = 118.92(7).
In diethyl ether solutions of freshly synthesized III, the 1H NMR and 13C NMR spectra show twice as much signals as in solutions that have been measured one or two days later. The doubling of signals is attributed to the presence of a second compound III′, which slowly disappears and transforms to III on standing in diethyl ether. Indeed, after a few days in diethyl ether solution, III′ is fully converted to III. So far, we have not been able to determine the molecular structure of this meta-stable compound III′, which is generally present in the reaction mixture in a higher ratio compared to the end-product III. Molecule III′ could be a diethyl ether adduct of III, which on prolonged standing steadily loses the coordinating base, or the C2-symmetric isomer of III (the two hydrogen atoms standing on the same side of the Al2O2 ring; see also Fig.1). This C2-isomer III′ could be energetically less favourable and could transform to the Ci-isomer III under the coordinating activity of diethyl ether.
Since the reaction between I and II occurs on the molecular scale, a homogenous aggregation of lead particles takes place, which imparts a strict control of the size distribution in the obtained colloidal solutions. The stabilization of clusters was investigated in both polar (tetrahydrofuran, acetonitrile, thiophene, thiophenol, and diethyl ether) and non-polar (toluene and n-hexane) solvents. The size of lead clusters in the solution depends on the temperature of the reaction medium and the concentration of the precursors. Scheme 2 shows the different steps involved in the synthesis of lead particles and their infiltration in porous aluminium-oxide membranes.

The reduction of I in highly concentrated reaction mixtures results in an uncontrolled particle growth responsible for the precipitation of polycrystalline lead powder as a black solid. A similar observation was made whilst performing the reaction at room or elevated temperatures. Nevertheless, lead colloids could be obtained when the reaction was performed at –100 °C with a slow addition of II. In contrast to the noble metal colloids, which are stable over large time periods, the lead colloids obtained in this study have a limited lifetime when oxygen- and sulphur-containing donor molecules are used to passivate the surface of the lead clusters. However, colloidal solutions stable up to several weeks were obtained by using acetonitrile as the coordinating ligand. In order to examine the effects of metal–donor interactions on the stability and size of lead colloids, solvents with different donor atoms (N, S, and O) were used. Acetonitrile-containing systems were found to be more stable than the others presumably due to the higher affinity of lead towards nitrogen when compared to sulphur and oxygen.
A photography of these colloidal solutions is shown in Fig. 2 . In view of the colour variations of the solutions obtained after the reduction of I (Fig. 2), UV–Vis absorption spectra of the colloids and of the starting materials were recorded and compared. In order to understand the chemical lead–ligand interaction in lead colloids, the absorption spectra of lead particles generated in oxygen- (tetrahydrofuran), sulphur- (thiophene) and nitrogen-containing (acetonitrile) solvent molecules were recorded (Fig. 3A ). The background due to the solvent absorption was removed from the experimental data shown in Fig. 3B-D. The spectra of the lead precursor and the in situ-formed by-product amido-alane were also recorded for reference. For clarity, four reference lines (a, b, c, and d) corresponding to the major spectral features were drawn in Fig. 3A. Among the three colloids, the features a and b dominate in Pb/THF and Pb/thiophene systems whereas the feature c is more prominent in the Pb/acetonitrile system. While Pb/THF (Pb/diethyl ether) and Pb/thiophene exhibit unstable green-coloured solutions, the Pb/acetonitrile solution results in a stable yellow colloid. The instability of the former two systems was studied by recording time-dependent UV absorption spectra (Fig. 3B and C). The decrease of the intensity of features b and d is concordant with the observed colour change. This phenomenon of disappearance of the green colour is also observed when the suspensions are exposed to sunlight or strong monochromatic light.

Photography of lead colloids showing the effect of solvent and concentration on the colour (A: THF; B–E: increasing concentrations of starting materials (12, 18, 30 and 60 mmol l–1) in acetonitrile as solvent.
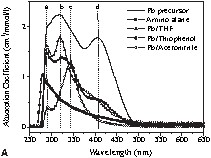
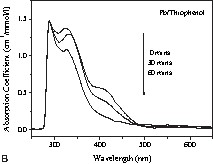
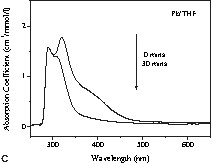

(A) Absorption spectra of Pb[N(SiMe3)2]2 (‘Pb precursor’), {[(Me3Si)2N](H)Al(OtBu)}2 (‘amino alane’) and Pb particles in different solutions. Time-dependent absorption spectra of Pb particles in (B) thiophenol and (C) THF show the instability of Pb colloids in the above solutions. (D) Absorption spectra of Pb colloids in acetonitrile show a gradual change in spectral weight due to the different concentrations of lead particles (C1: relative concentration of Pb[N(SiMe3)2]2, C2: relative concentration of (tBuO–AlH2)2 before reaction).
In a different experiment, a systematic variation in the concentration of lead precursor revealed a distinct colour difference emerging either from the difference in the average particle size or from the degree of aggregation of the lead clusters present in the solutions. In order to investigate the size–colour dependence, optical absorption spectra (Fig. 3D) of the different solutions were recorded. The spectra are normalized to the concentration and for comparison purposes. The systematic shift of the absorption peak with increasing concentration towards larger wavelength indicates the presence of larger lead particles in the case of higher concentrated solutions. Apparently, a larger number of nuclei are generated in the concentrated solutions, which increases the probability of inter-particle collisions and coalescence, responsible for the particle growth. However, this presumption is not supported by TEM studies, which do not show any significant particle growth in going from sample B to E (Fig. 2). The theory of Mie scattering [35–37] from similar small aggregate clusters suggested that the plasmon resonance absorption of the aggregates would have an additional long wavelength peak relative to the plasmon resonance from dispersed nanoparticles. This feature arises from the interactions of the plasmon resonances of the individual metal particles. Another potential explanation for the colour changes is the effect of different degrees of aggregation, which can usually give rise to a double-peak-like structure due to plasmon–plasmon interaction in aggregates of metal nanoparticles. The plasmon resonance of individual lead nanoparticles (av. size = 10 nm) is observed at 218 nm [37]. This feature is not observed in our samples due to the recorded spectral range (250–650 nm).
The phase characterization was performed by X-ray diffraction analysis of the powder obtained by drying a colloidal solution in vacuum (~ 10–3 mbar). The data obtained are shown in Fig. 4 and correspond (PDF: [4-686]) to the cubic phase of lead metal. Due to the small particle size and large surface area, the lead particles are highly susceptible to atmospheric oxidation and can be completely oxidized under aerobic conditions to nano-crystalline lead oxide (Fig. 4). The formation of lead oxide was also confirmed by the FT–IR spectrum that exhibits a peak at 560 cm–1 due to the Pb–O stretching frequency.
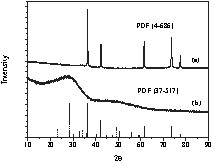
XRD patterns (Cu Kα radiation) of (a) the Pb particles obtained from Pb/acetonitrile system and (b) the solid obtained after the aerial oxidation of Pb nanoparticles. The solid and dashed vertical lines represent the reference Pb (PDF[4-686]) and α-PbO2 (PDF[37-517]), respectively [38].
The lead particles in the colloidal solutions were examined by transmission electron microscopy, which showed that the particles obtained in acetonitrile (Fig. 5A ) medium were significantly smaller than those obtained in tetrahydrofuran (Fig. 5B). The average particle-size in acetonitrile and THF solutions were found to be ca 10 and 35 nm, respectively. The particle size is highly homogenous, as observed in the scanning electron micrographs of larger lead clusters aggregated on the surface of alumina membranes (Fig. 6 ). The spot-EDX analysis performed randomly on different particles confirmed them to be lead metal (Fig. 6C). To fill porous alumina membranes with lead nanoparticles, a membrane of pore size 60 nm was treated with the colloidal THF solution through vacuum induced diffusion. However, it was not possible to fill the pores due to the mismatch between the particle and the pore sizes and the particles were found to conglomerate on the membrane surface. A typical aggregate structure is shown in Fig. 7A , which reveals the regularity of the cluster size and shape existing in the solution. The average particle size is ca 200 nm, which is significantly larger than the pore size of the membrane. When colloidal solutions containing a smaller concentration of lead clusters were used to impregnate the membrane, individual particles were found to be present on the pores of the membrane. The distribution was not uniform, due to the reduced density of lead clusters in the solution. For comparison, a non-treated membrane is shown in the inset of Fig. 7A. Although it was possible to fill the membrane with smaller particles, the cross-sectional SEM investigations of alumina membrane (Fig. 7B) revealed that very few lead particles were present in the channels of the membrane. It is possible that the metal particles, which are loosely held in the pores, are blown away when the membrane is cleaved for the microscopic analysis.
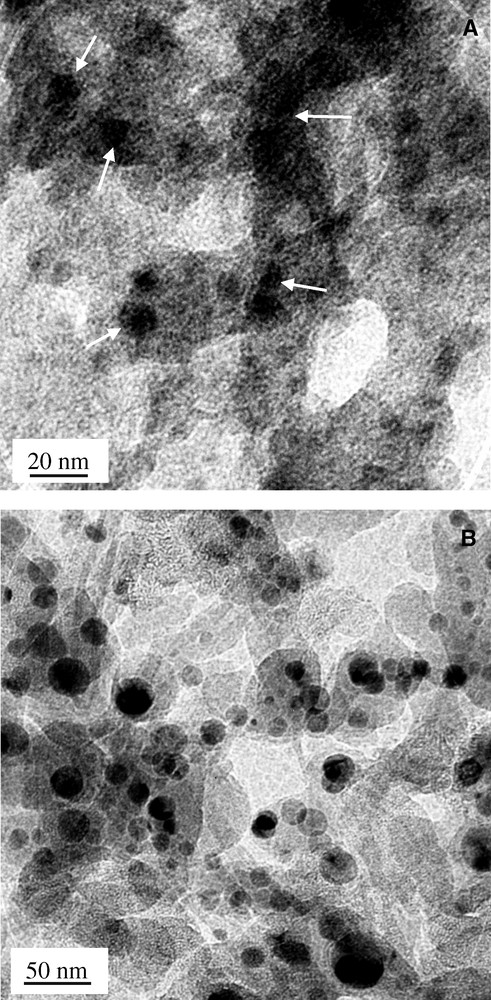
Transmission electron micrographs of Pb particles prepared in (A) acetonitrile and (B) THF solvents.

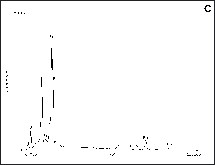
(A-B) Scanning electron micrographs of Pb particles agglomerated on the membrane surface and (C) the corresponding EDX spectrum; Cu peaks are due to the substrate.

Scanning electron micrographs of alumina membranes obtained after (A) vacuum-induced infiltration of Pb particles and (B) reduction of lead amide within the pores of the membrane (cross-sectional).
An alternative approach to fill the membranes consists of an in situ reduction of the lead precursor within the pores. However, the particle size was not homogeneous possibly due to the gradient in the chemical reaction occurring in the nano-channels. Moreover, the surface protection of nanoparticles generated in the pores seems to be less effective, which leads to an uncontrolled particle growth. Further experiments to optimise the fabrication of Pb/Al2O3 systems are currently under progress.
4 Conclusions
Lead colloids containing nano-sized metal particles of different diameters have been obtained by a redox reaction between a molecular lead precursor and a tert-butoxy derivative of aluminium hydride, used as a reducing reagent. The transformation of the lead amide to metallic lead is a chemically controlled process, which allows a precise control over the size of the resulting lead clusters either by changing the concentration of the reactants or the donor solvent. Colloidal solutions showing yellow, bright yellow, orange and burgundy red colours were produced by gradually increasing the concentration of lead particles in the solutions. The colour variations seem to be a collective effect of the different particle sizes and degrees of aggregation. The increase in particle size is observed by transmission electron microscopy, while evidence for aggregate formation is obtained by a feature observed towards the longer wavelength in the optical absorption spectra corresponding to plasmon–plasmon interaction in conglomerated metal clusters. The preliminary results indicate that the nature of the metal–ligand interaction is responsible for the stability of the colloids as well as the particle growth. For instance, smallest particle size and long-time stability (several weeks) of the colloidal solution was achieved when using acetonitrile (N-containing donor) as the surface-passivating medium. On the other hand, oxygen- (THF or diethyl ether) and sulphur-containing (thiophenol) ligands are not effective in shielding the surface of the nanoparticles, apparently due to the higher steric profiles of the organic molecules in comparison to linear acetonitrile molecules and the nature of the donors. As a result, Pb/THF and Pb/thiophenol systems show a phase separation, after ca 2 days, into the continuous medium (solvent) and a precipitate that was confirmed to be fine lead powder that could be redispersed in the solvent by ultrasonic treatments. A part of the study was focused on the synthesis of ordered lead structures by aligning the lead nanoparticles in the pores of aluminium-oxide membranes. The infiltration of lead clusters in the pores of alumina membrane was attempted either by (i) vacuum-induced diffusion of lead particles from a colloidal solution or (ii) by reducing the lead amide within the pores of the membrane. The vacuum-induction method was not successful because of the size of the lead particles (blocking of the pore openings). The in situ-reduction approach was found to be more useful and produced a better distribution of lead clusters in the channels. However, the reaction gradient led to particles of different sizes.
Acknowledgements
Authors thank the ‘Deutsche Forschungsgemeinschaft’ for providing financial support in the framework of priority research programme Halbleiter- und Metallcluster als Bausteine für organsierte Strukturen (SPP 1072) as well as the ‘Fonds der Chemischen Industrie’. Catherine Cazin is acknowledged for kindly reading the manuscript.