

1 Introduction
The noxious effects on human health of nitrosation and nitration processes targeted to bioactive molecules have been known since a long time. However, after the discovery in 1987 of nitrogen monoxide (nitric oxide, NO) as a major endogenous mediator of physiological control and a critical amplifier of cytotoxic processes, there has been a considerable burst of interest in the chemistry of biological nitrosations and nitrations. The present account aims to provide a concise overview of the latest achievements in this expanding area of research, as resulting from the continuing efforts in the author's laboratory and other research centers. Because of space restrictions, coverage of the topic will be illustrative rather than comprehensive, and the interested reader is referred to the relevant literature and the many excellent reviews in the field.
2 Origin, sources and pathophysiological roles of nitrosating and nitrating species
One of the major exogenous sources of nitrosating species is represented by nitrite ions which are found at μM levels in cured or pickled meats, vegetables, fertilizers and polluted drinking water. In the acidic environment of the stomach (pH 2.5–3 during digestion), nitrite is converted to nitrous acid (pKa = 3.2) which is readily decomposed to give a range of nitrosating species, including nitrosonium ion (NO+) and dinitrogen trioxide (N2O3). These species can promote the nitrosative deamination of primary aromatic amines, including purines and pyrimidines. Deamination of cytosine, adenine, or guanine results in the formation of uracil, hypoxanthine, and xanthine in that order. Conversion of cytosine and methyl cytosine can lead ultimately to a base pair substitution mutation, while deamination of adenine and guanine results in transversion mutations. Moreover, the instability of hypoxanthine and xanthine in the DNA structure leads to rapid depurination and consequent single strand breaks. Nitrosation of secondary aliphatic and aromatic amines can also produce potentially carcinogenic nitrosamines. Such nitrosamines, like many chemical carcinogens, are thought to promote mutagenesis and carcinogenesis via their ability to alkylate specific sites in DNA following metabolic activation.
Other sources of toxic nitrosating and nitrating species include combustion, industrial processes, environmental tobacco smoke and exhaust fumes from cars, which produce relatively high levels of nitrogen oxides, including nitrogen dioxide (NO2). This latter is a ubiquitous and toxic contaminant of urban air in industrialized countries, which has been implicated in the adverse effects of smoggy air on the respiratory organs.
Yet, one of the leading species responsible for nitrosation and nitration processes in vivo is nitrogen monoxide (commonly referred to as nitric oxide, NO), a signaling mediator generated by the endothelium to maintain vascular homeostasis through the regulation of blood pressure and leukocyte-platelet activation [1–4]. The importance of NO as a physiological mediator is highlighted by the awarding of the Nobel Prize for Physiology and Medicine in 1998 for description of the interaction between NO and the haem group of soluble guanylyl cyclase.
Besides being the critical mediator of smooth muscle relaxation, NO is involved in a number of neuronal functions including long-term potentiation (a cellular basis for learning), synapse formation, drug tolerance and dependence, modulation of sensory and motor pathways, local regulation of cerebral blood flow, neuroendocrine regulation, learning, and feeding and sexual behavior.
NO is synthesized in leaving organisms by the action of a calcium/calmodulin-dependent oxygenase, nitric oxide synthase (NOS) [5], which oxidizes a guanidinium nitrogen of L-arginine to yield NO and l-citrulline (Fig. 1).
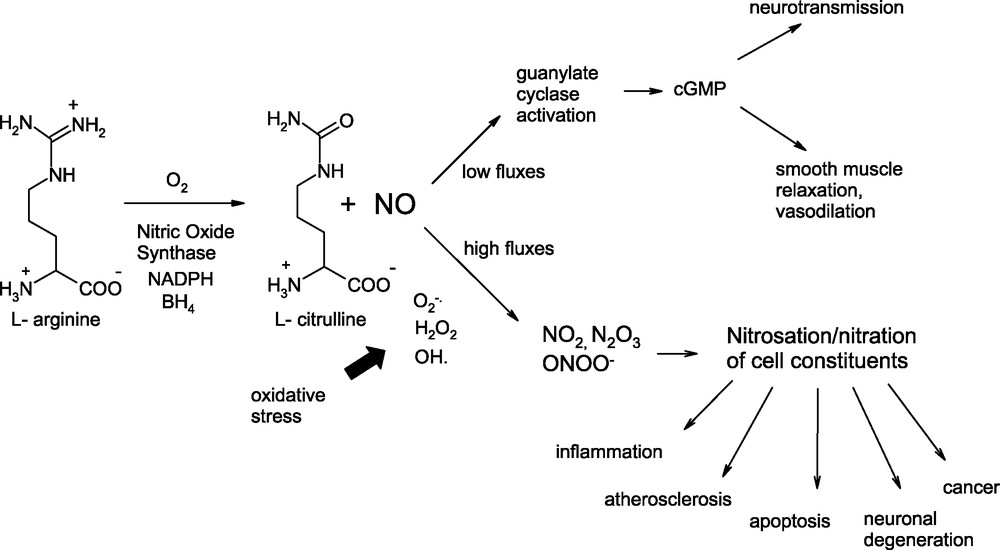
Synthesis and dual role of nitric oxide (NO) in living organisms.
Although NO subserves a variety of positive roles in human physiology, its aberrant generation under oxidative stress conditions is now recognized to be associated with a series of adverse events, such as inflammation, atherosclerosis, apoptosis, neuronal degeneration and cancer [6,7]. These latter are thought to be mediated by a range of nitrosating and nitrating species produced by oxidative conversion of NO, including NO2, N2O3, and peroxynitrite (ONOO–), the highly reactive nitrating agent produced by coupling of NO with superoxide [8]. Peroxynitrite was initially regarded as the primary determinant of biological nitrations, especially in the presence of CO2 [9–11], but it seems now clear that several pathways, not mutually exclusive, may actually be involved [12].
One of the most important targets of nitrating agents are tyrosine residues in proteins. The reaction involves one electron oxidation of the phenol ring to give a phenoxyl radical which couples with NO2 to form 3-nitrotyrosine (Fig. 2).
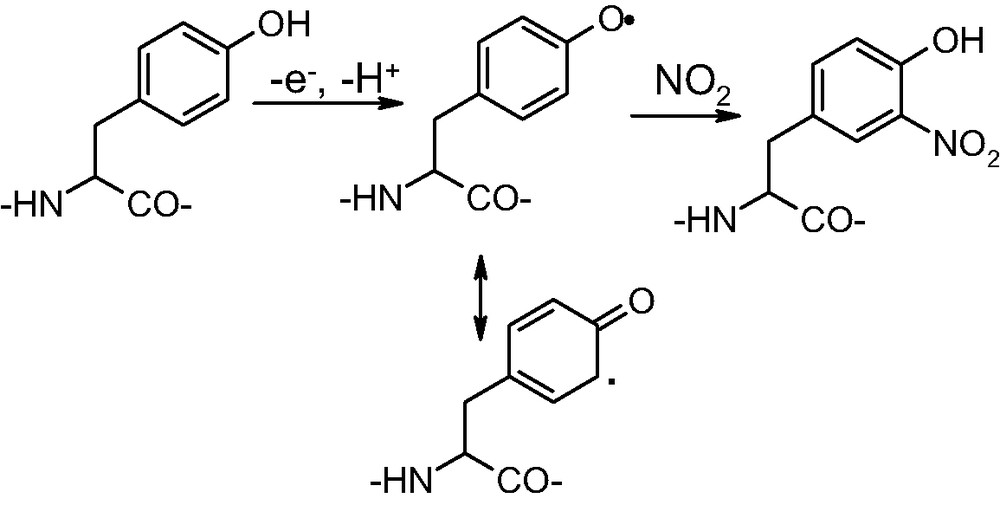
Tyrosine nitration in proteins.
3-Nitrotyrosine formation is now an established mechanism of post-translational protein modification which is often associated, but not causally linked, with deleterious effects, such as loss of structural integrity, enzyme inactivation, receptor modification and enhanced proteolysis. In view of the very large number of papers dealing with protein tyrosine nitration, the reader is referred to the extensive literature in the field [13–29].
3 Polyunsaturated fatty acids as targets of nitrosating and nitrating species
Several studies carried out in our laboratory and other centers have shown that, in addition to proteins, other biomolecules can be targeted by nitrosating and nitrating species. These include nucleic acids [30–32], thiol compounds [33–37], estrogens [38], tocopherols [39,40], retinoids [41], as well as polyunsaturated fatty acids and catecholamines. These latter will be the main focus of the present review.
The importance of polyunsaturated fatty acids as potential targets of nitrosating and nitrating species in vivo has recently been demonstrated by the identification of nitrated linoleate in human plasma [42,43]. Nitrolinoleates have been shown to inhibit neutrophil functions and platelet aggregation, and to stimulate smooth muscle relaxation [44–47]. The platelet inhibitory actions of nitrolinoleates indicate that nitration reactions that occur following NO generation in an oxidizing environment can alter the activity of lipids, and throw light on potential mechanisms by which NO-derived species may modulate the progression of vascular injury.
A first chemical insight into the nitrosation and nitration of polyunsaturated fatty acids was gained in 1996 when the reaction of ethyl linoleate with nitric oxide in air-equilibrated cyclohexane was investigated [48]. Product analysis revealed a bewilderingly complex mixture of species, some of which could be isolated and identified as regio- and stereoisomeric nitroalkene and nitronitrate derivatives by extensive 2D-NMR and GC–MS analysis. It should be noted that no reaction occurred in the absence of oxygen, consistent with the established inertness of NO toward alkenes.
In an extension of that study [49] it was found that the chemical course of the reaction of polyunsaturated fatty acids with NO changed considerably with the medium: whereas in cyclohexane it afforded mainly nitration products, in aqueous phosphate buffer at pH 7.4, it led to the formation of peroxidation products (Fig. 3).
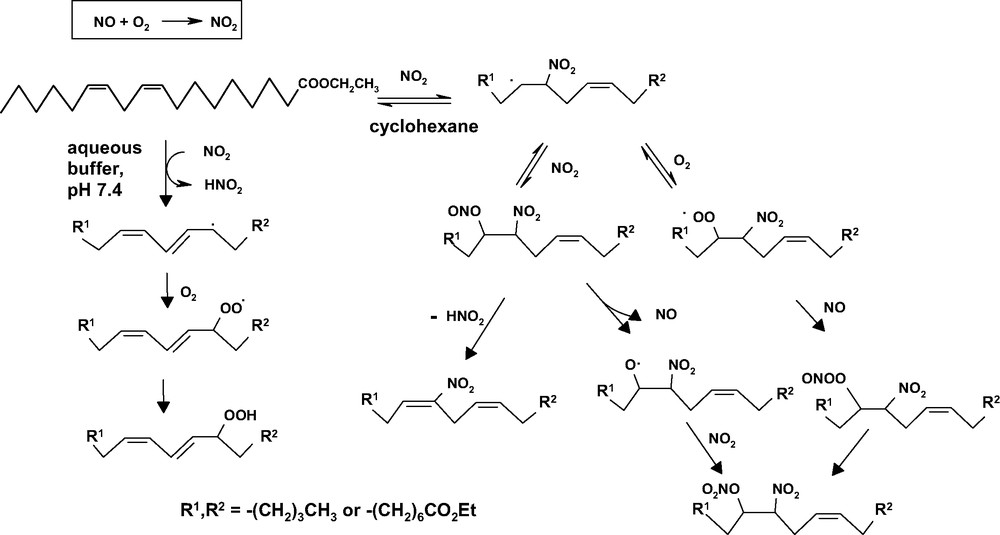
Medium-dependent reaction pathways of ethyl linoleate with NO in the presence of oxygen.
This effect can be explained considering that in cyclohexane the NO2 generated by oxidation of NO can add to the double bonds by a fast and reversible process to form β-nitroalkyl radicals. These latter can be trapped by NO2 or oxygen to give eventually nitration products. However, in aqueous medium, within the pH range 2–13, the solubility of NO is 1.8 × 10–3 M, i.e. one order of magnitude lower than in cyclohexane, and trapping of the β-nitroalkyl radical would be less efficient. As a result, irreversible H-atom abstraction by NO2 becomes prevailing.
In addition to NO/O2, ethyl linoleate has been shown to react efficiently with nitrite ions in an acidic biphasic medium consisting of cyclohexane/1% aqueous sulfuric acid [50]. Flash chromatographic separation of the mixture followed by repeated TLC fractionation led to the isolation of ethyl 10-nitrolinoleate and 12-nitrolinoleate in pure form, along with 9-nitro and 13-nitrolinoleates, an unusual allylic nitroderivative, a dinitroderivative (regioisomers) and a mixture of isomeric 2-nitroalcohol derivatives. A plausible mechanistic scheme leading to the reaction products is reported in Fig. 4.
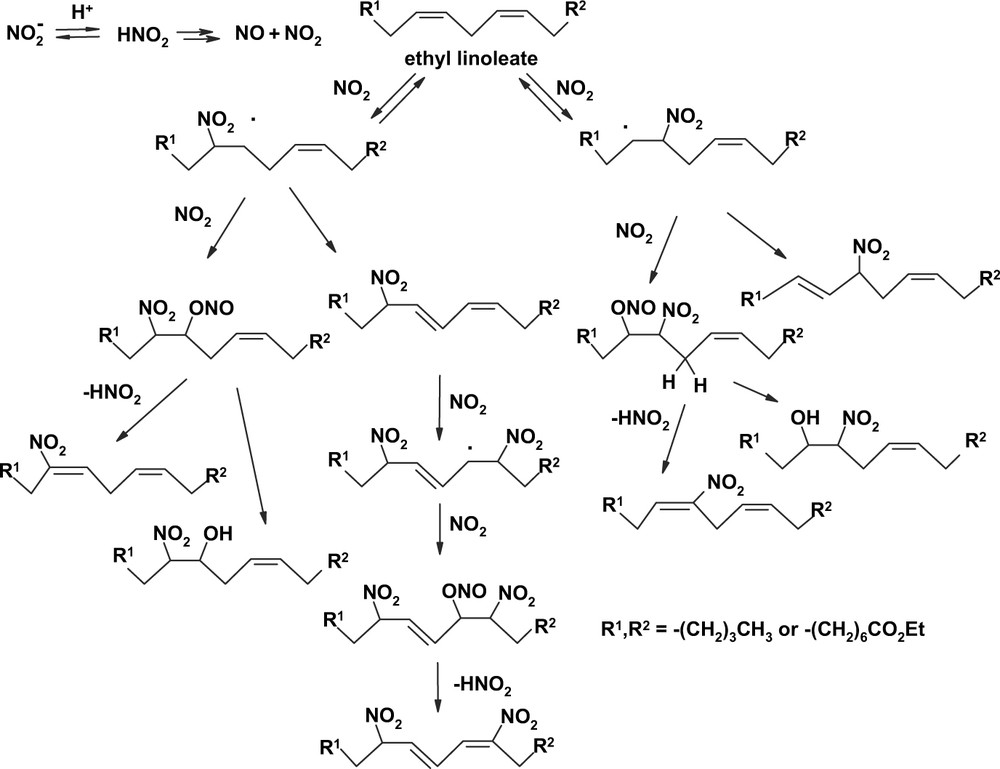
Mechanism of reaction of ethyl linoleate with nitrite ions in acidic medium.
The first step is the attack of NO2 produced by decomposition of nitrous acid to give β-nitroalkyl radicals. These can partition between H-atom abstraction, leading to allylic nitro derivatives, or coupling with NO2 at the oxygen atoms, leading eventually to nitroalkene or 2-nitroalcohol derivatives via nitrous acid elimination or hydrolysis.
A deeper insight into the nature of the products formed by reaction of ethyl linoleate with acidic nitrite was gained by an integrated approach based on the use of 15N-labelled nitrite and extensive GC–MS and 2D NMR (including 15N NMR) analysis of the crude or partially fractionated mixture [51].
In a related study [52], the attention was focused on the reaction of nitrite ions with an important metabolite of linoleic acid, namely (9Z,11E,13S)-13-hydroxyoctadeca-9,11-dienoic acid (13-HODE). The reaction was carried out on a suspension of the lipid in phosphate buffer at pH 3–5.5 and resulted in the formation of two main nitrated derivatives, one of which proved to be an unusual breakdown product featuring a 4-nitro-2-oximino-3-butenal moiety.
This product was proposed to originate from the initial addition of NO2, produced by decomposition of nitrous acid, to the 9-position of 13-HODE to give a delocalized radical intermediate which can couple with another molecule of NO2 to give a transient nitronitrite derivative. This would undergo breakdown with loss of nitroxyl to give hexanal and a nitroalkenal derivative in equilibrium with an isomer displaying a reactive methylene group. This latter would be susceptible to nitrosation to give eventually the α-oximinoaldehyde product (Fig. 5).
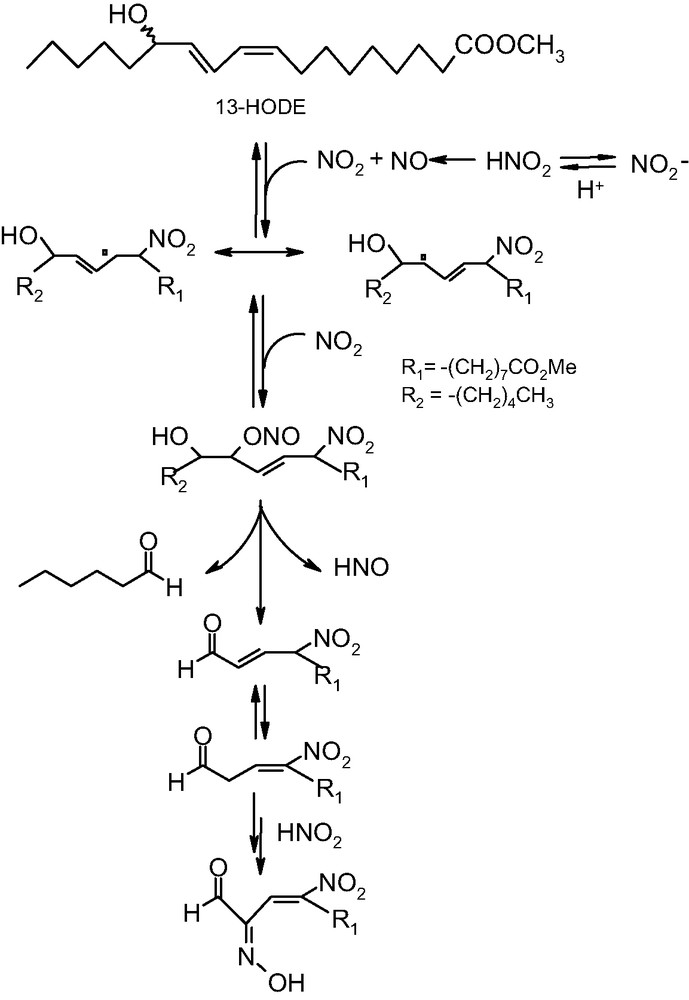
Suggested mechanism of breakdown of 13-HODE induced by nitrite ions in acidic medium.
4 Catecholamines as targets of nitrosating and nitrating species
The formation of nitrated catecholamines in vivo was first demonstrated in 1996 by a Japanese group reporting the occurrence of 6-nitronoradrenaline in mammalian brain in levels proportional to nitric oxide synthase activity [53]. 6-Nitronoradrenaline was found to inhibit catecholamine reuptake and catechol-O-methyltransferase activity.
The first evidence about the susceptibility of catecholamines to nitration was obtained in 1994 when De la Breteche et al. [54] reported the formation of nitrocatecholamines by reaction of catecholamines with NO at physiological pH. The reaction was found to depend on the presence of oxygen and proceeded with high regioselectivity at the 6-position [55]. In a subsequent study [56] several aspects of this chemistry were addressed and a mechanism was proposed, involving one-electron oxidation of the catecholamine by NO2, generated by oxidation of NO, followed by radical coupling of the semiquinone with NO2 or with NO (Fig. 6).
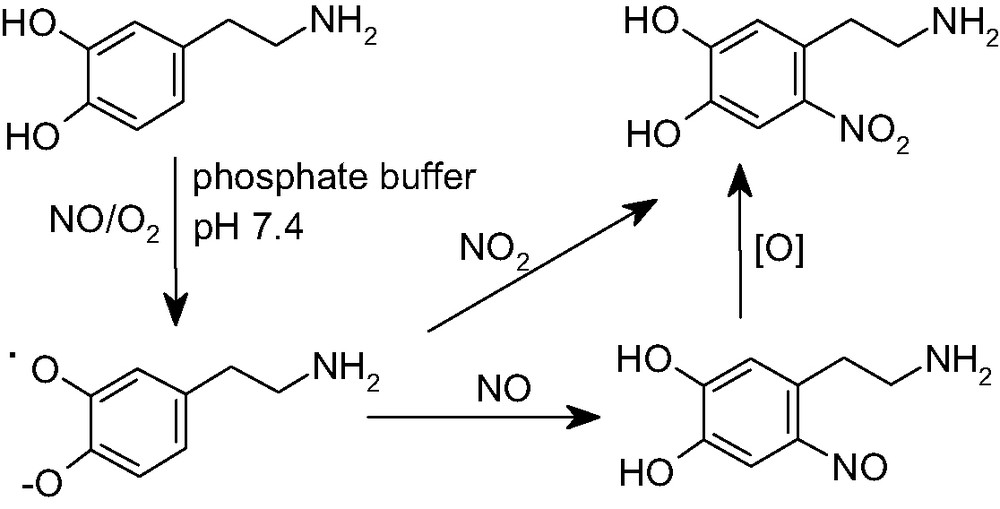
Generation of 6-nitrodopamine by reaction of dopamine with NO in the presence of oxygen.
More recently, an alternative mechanism has been proposed for catecholamine nitration involving electrophilic attack by N2O3 followed by an oxidation step [57].
In another study [58], additional potential routes for catecholamine nitration were highlighted, involving oxidation of the catecholamine by peroxidase/H2O2 or the Fenton reagent (Fe2+/EDTA/H2O2) in the presence of nitrite ions (Fig. 7).
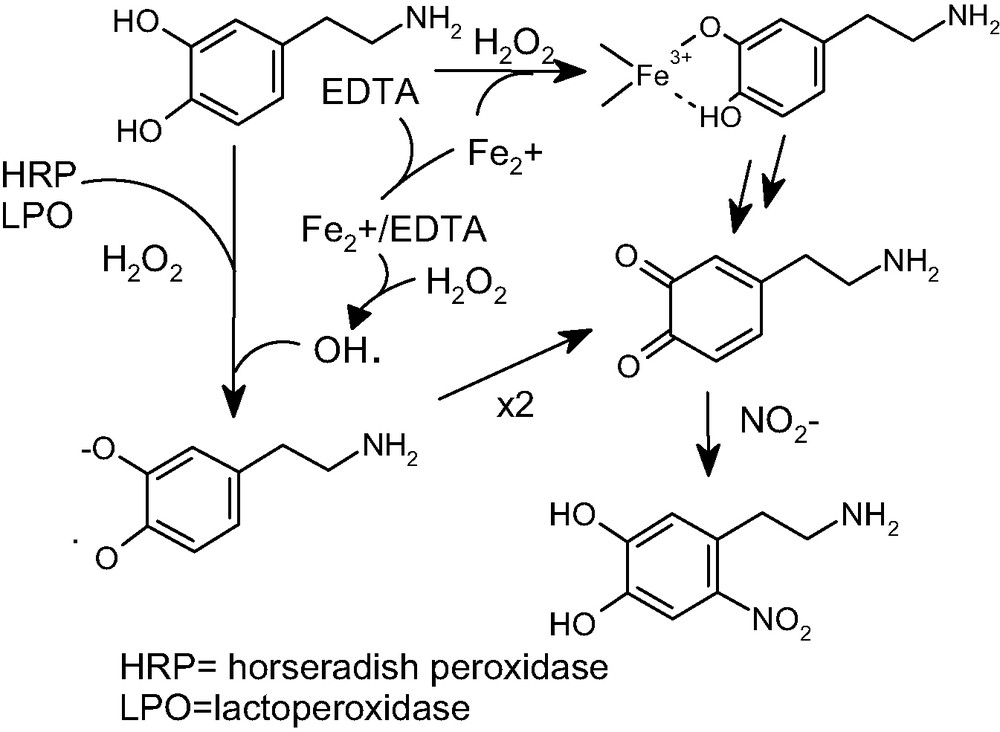
Generation of 6-nitrodopamine by oxidation with hydrogen peroxide-based systems in the presence of nitrite ions.
Under these conditions, nucleophilic addition of nitrite ions to dopamine quinone is the most likely mechanism. In line with this view is the finding that quinone-trapping reagents can inhibit the reaction, leading to conjugation products, and that tyrosinase, a copper containing enzyme which catalyzes two-electron oxidation of catechols to o-quinones, in the presence of nitrite ions gives rise to 6-nitrodopamine in comparable yields.
Since catecholamine nitration occurs in oxidizing environments [59], which would make oxidation of the resulting nitrocatecholamines a likely event, in a separate study the oxidative behavior of 6-nitrodopamine was investigated [60,61]. Oxidation with peroxidase/H2O2 led to the formation of two main isolable products that were identified as regioisomeric dimers in which one unit of 6-nitrodopamine was linked through an ether bridge to a nitroindoline moiety. A plausible mechanism of formation of these dimers is shown in Fig. 8.
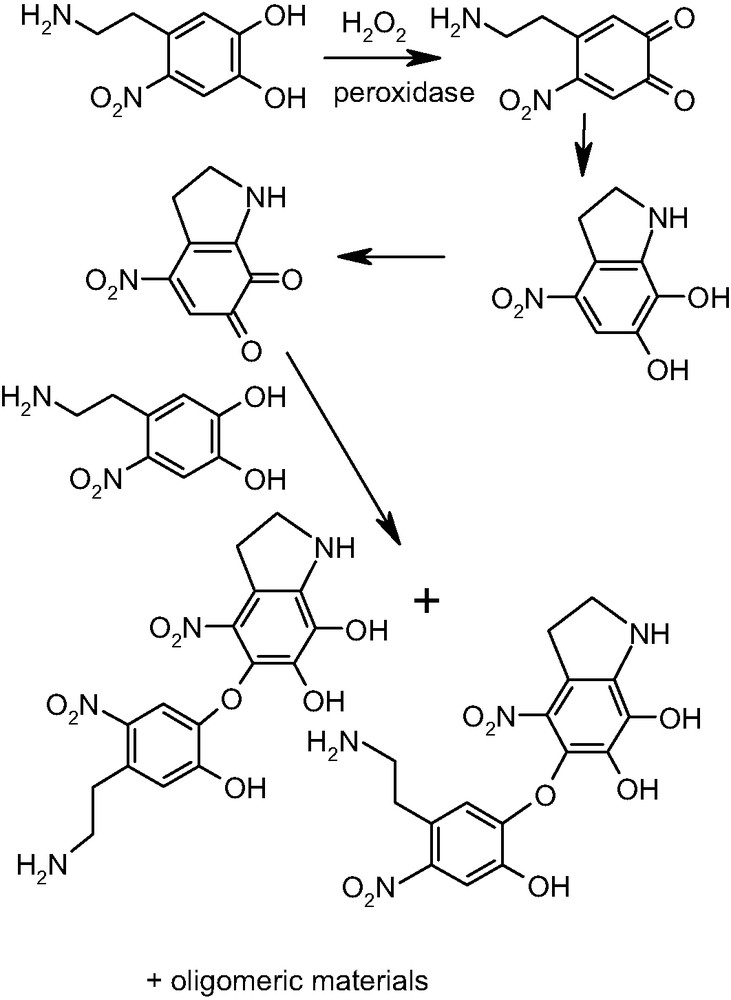
Oxidative conversion of 6-nitrodopamine to dimers.
Interestingly enough, 6-nitrocatecholamines proved to be effective competitive inhibitors of neuronal NOS [62] interfering both with substrate binding and tetrahydrobiopterin-induced enzyme dimerization and activation. It was suggested that 6-nitrocatecholamines interact with the l-arginine binding site near the haem prosthetic group through the nitrocatechol moiety, and affect tetrahydrobiopterin binding through the aminoethyl side chain. This conclusion was supported by the observation that nitrocatechols lacking the aminoethyl chain do not exhibit tetrahydrobiopterin antagonizing properties [63].
5 Mechanism of action of putative cancer chemopreventive agents with antinitrosating properties
Caffeic acid and chlorogenic acid, which are found in relatively high levels in coffee, tea, fruits and cereals, are commonly ascribed potent antinitrosating properties [64], and have been suggested to play an important role in prevention of toxic nitrosation reactions in the stomach following elevated nitrite intake. However, the chemistry underlying their mechanism of action remained poorly understood.
In a systematic examination of this issue, it was found that at low concentrations and under conditions simulating those within the stomach, caffeic acid reacts with nitrite ions to give a complex mixture of products including mainly an α-hydroxyoxime, an α-methoxyoxime (reflecting the presence of methanol in the mixture as cosolvent), 3,4-dihydroxybenzaldehyde and small amounts of a benzoxazinone derivative [65,66]. At higher substrate and nitrite concentrations, the main products were, besides the hydroxyoxime, an α-ketooxime and a furoxan derivative (Fig. 9).
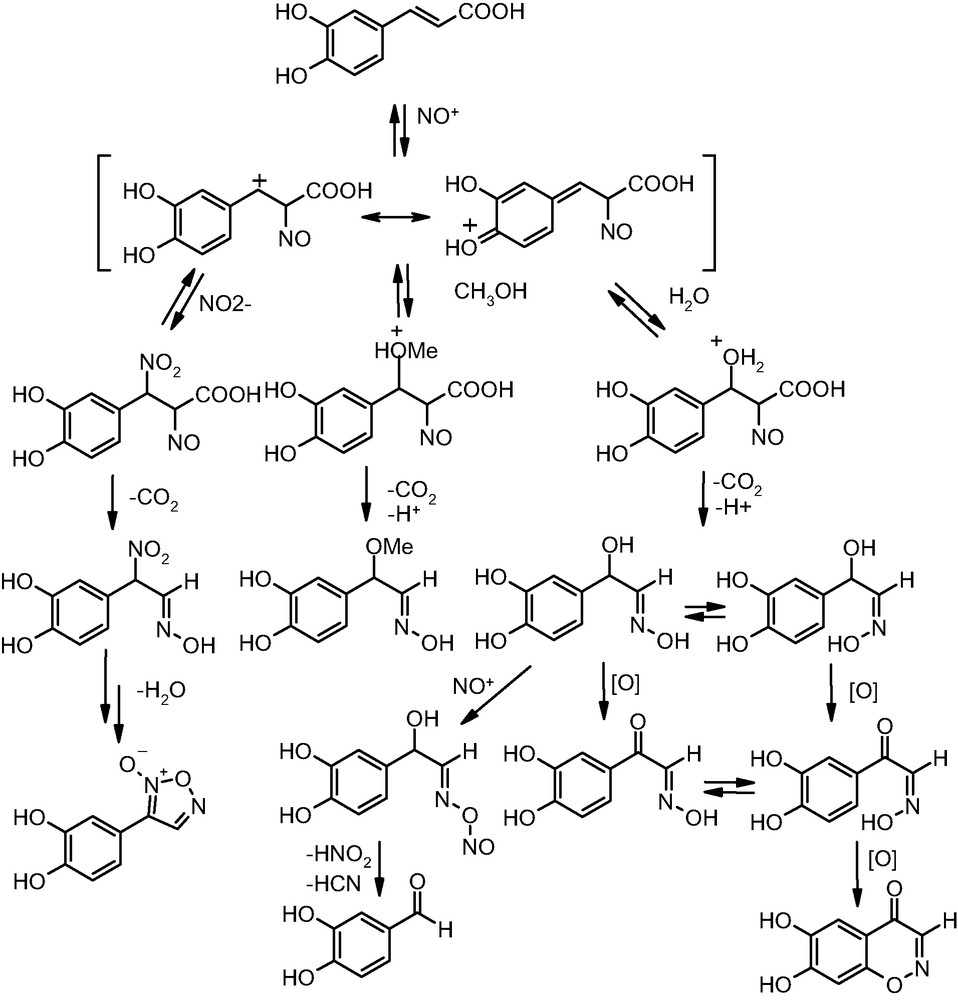
Proposed mechanism of formation of oxime, aldehyde, furoxan and benzoxazinone products by reaction of caffeic acid with nitrite ions in acidic medium.
Under the same conditions, both caffeic acid methyl ester and chlorogenic acid gave mainly ring nitration products. The different reactivity displayed by caffeic acid and its esters on exposure to nitrite ions in acidic medium can be accounted for in terms of competitive pathways whose outcome is dictated by the availability of the free carboxylic acid functionality.
In the case of caffeic acid, the first step is likely to be the attack of nitrosonium ion to the 2-position of the propenoate chain to give a nitroso adduct. This nitroso intermediate would bear a positive charge and could be attacked by the main nucleophiles in the medium, namely water, to give the hydroxyoxime, 3,4-dihydroxybenzaldehyde, the ketooxime and the benzoxazinone; methanol, to give the methoxyoxime; and nitrite ions, to give eventually the furoxan. In all of these pathways, the driving force would be provided by decarboxylation of the adduct with isomerization of the nitroso group to the oxime function (Fig. 9).
In the case of caffeic acid esters, the initial nitrosation intermediate can not evolve through the decarboxylation channel and reverts, so that ring nitration eventually becomes favored.
In the presence of glutathione, the most prevalent intracellular thiol antioxidant and a potential anticarcinogen deputed to mobilization of xenobiotic toxicants, the reaction of caffeic acid with nitrite ions in acidic medium takes a different course, to give four novel products that were identified as stereoisomeric α-glutathionyloxime derivatives [67] (Fig. 10). These arise by a mechanism akin to that proposed for the other oxime products.
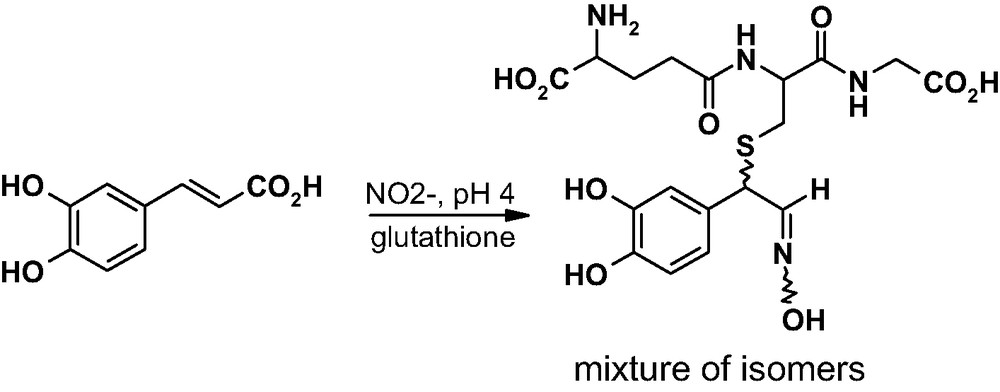
Main products formed by nitrite-mediated reaction of caffeic acid with glutathione.
In the case of chlorogenic acid, the same reaction led to the formation of a series of conjugation products, including notably a triadduct [68] (Fig. 11). Since oxidation of chlorogenic acid with tyrosinase in the presence of glutathione gives the same adducts in comparable yields, it seems likely that in acidic medium nitrite ions bring about catechol oxidation to give the quinone which undergoes nucleophilic attack by the thiol.
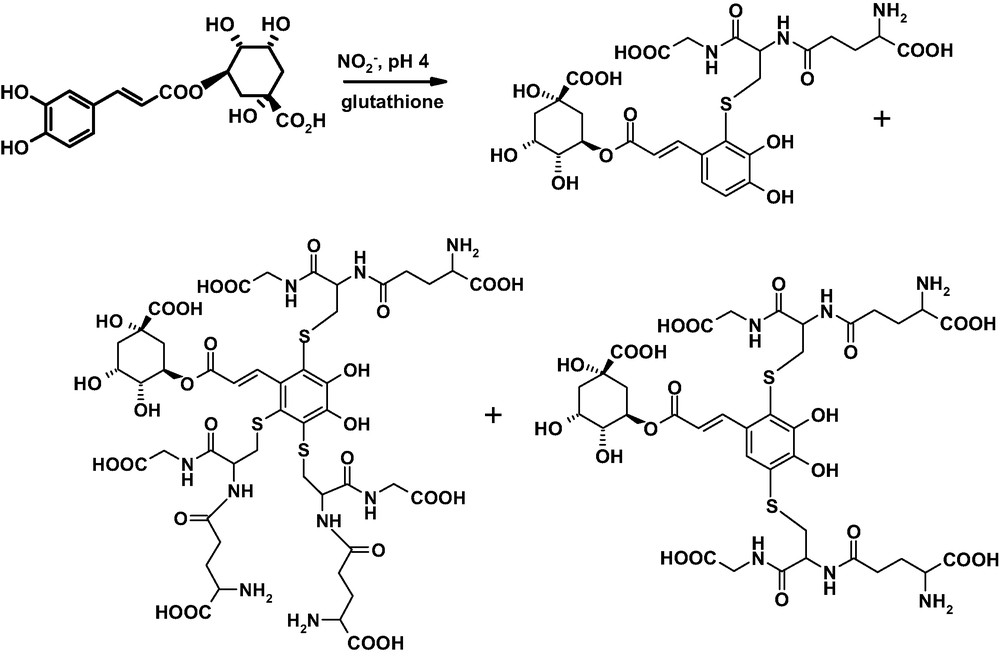
Main products formed by nitrite-mediated reaction of chlorogenic acid with glutathione.
6 Conclusions and perspectives
The present account has provided some examples of reactions of nitrosating and nitrating species of biomedical relevance with potential target molecules. The chemistry emerging from these studies clearly shows that the (patho)physiological actions of these species, be they of exogenous or endogenous origin, are rooted in their capacity to covalently modify biological macromolecules, a process which may ultimately lead to altered biomolecular functions. Likewise, protective mechanisms rely on the activity of molecules usually referred to as ‘antinitrosating’ (in close analogy with the more familiar case of antioxidants) which act as defences whenever an aberrant production of toxic nitrosating and nitrating species occurs. Further insights into the roles these species play in pathological states will depend on a closer understanding of their reaction behavior and targets. Pursuit of this goal will hopefully disclose novel therapeutic strategies and/or modalities for prevention of disease.
Acknowledgements
Research in the author's laboratory was carried out in part with financial support of Italian Ministry of University (MIUR, projects PRIN 2001 and 2003) and Ministry of Health. The assistance of Mrs. Silvana Corsani is gratefully acknowledged.