

1 Introduction
Of all neutral parent carboranes known, the cage of icosahedral para-carborane 1,12-C2B10H12 1 is the most robust after 1,10-C2B8H10 in terms of high thermal and chemical stabilities, and only degrading under extreme basic conditions [1]. The robustness of the cage is largely attributed to its much discussed ‘three-dimensional aromaticity’ [2]. However the terminal hydrogens of the para-carborane cage can easily be replaced by a variety of organic and inorganic substituents resulting in a diverse derivative chemistry [3,4]. As the cage in para-carborane has a five-fold symmetry through the axis of the cage carbons and the hydrogens at the cage carbons are easily substituted, there has been much interest in this carborane as a linker/building block for liquid crystalline materials [4–6], rigid rods [7] and materials with large hyperpolarisabilities [6,8–10].
Many experimental and theoretical studies on para-carborane derivatives have shown little or no π-conjugation between the cage and an unsaturated organic group, such as aromatic rings [10,11], ethynyls [6,12,13] and nitriles [12], or an anionic borane cage, B12H112– [9]. However recent carbon-13 NMR and UV–vis studies of various 1,12-diaryl-para-carboranes with organic electron donor and acceptor substituents show evidence of electronic transmission via the para-carborane cage [14]. The rate of hydrolysis of C-para-carboranylbenzyl toluene-para-sulphonates, where organic electron acceptors and donors are present at the second cage carbon, has been shown to depend on the acceptor/donor substituent giving further support for the transmission of electronic effects via the carborane cage [15]. The cyclic voltammetry response of the para-carborane complex with a CpFe(CO)2 group at each cage carbon is characterised by two discrete one-electron oxidations, clearly indicating transmission of electronic effects via the carborane cage, and similar conclusions could be drawn by comparison of the spectroscopic properties of this complex with those of closely related complexes [16].
Cobalt octacarbonyl, Co2(CO)8, and phosphine-substituted derivatives such as Co2(CO)6(μ-dppm), readily coordinate to alkynes RC≡CR′ through the displacement of two carbonyl ligands to give tetrahedral Co2C2 clusters of general form Co2(μ-RC2R′)(CO)4(L2) (L = CO, phosphine). The electrochemical response of the hexacarbonyl derivatives is characterised by a one-electron, diffusion-controlled reduction, which is often complicated by subsequent fast chemical reactions arising from cleavage of the Co–Co bond [17]. The chemical reversibility of the reduction process is improved through the presence of electron-withdrawing or bulky groups on the alkyne [18]. The introduction of phosphine [19] or phosphite ligands [20] results in the observation of a one-electron oxidation process within the electrochemical window of solvents such as CH2Cl2 and THF. The oxidation becomes more chemically reversible with increasing number of phosphite ligands, or through the use of bis-(diphenylphosphino)methane (dppm).
As we were interested in the mechanism by which electronic effects are transmitted through the para-carborane cage, here we synthesised a para-carborane cobalt cluster system with a Co2(μ-RC2R′)(CO)4(μ-dppm) substituent at both carbons of the para-carborane cluster. This compound was investigated by a combination of voltammetry and DFT-based computational work, and the electronic structures of redox-active cobalt clusters spanned by a 1,12-C2B10H10 bridging moiety were examined. These results are compared with the properties of systems featuring more conventional 1,4-C6H4 and C≡CC≡C (C4) bridging moieties. Part of this work has been communicated elsewhere [21].
2 Results and discussion
2.1 Synthesis and characterisation
The reaction of the mono-ethynyl carborane 2 [22] with Co2(CO)6(μ-dppm) in refluxing benzene afforded dark red 4 (40%), which was isolated from the reaction mixture by preparative TLC (Scheme 1). The IR ν(CO) spectrum contained terminal ν(CO) bands between 2077 and 1977 cm–1 in addition to the ν(BH) bands centred at 2656 cm–1, while in the 1H NMR the methylene protons of the dppm ligand were observed as partially resolved resonances centred at 3.27 and 3.40 ppm. The negative ion ES-mass spectrum of a solution in methanol containing NaOMe was characterised by isotopic envelopes from the [M–H]– and [M–H–CO]– ions [23]. We note here that 4 is not the first mixed carborane/organometallic dicluster of its kind; the complex [Co2(CO)6](μ-HC2CB10H10CH) has been prepared from 1-ethynyl-ortho-carborane and Co2(CO)6 but was not structurally characterised [24].
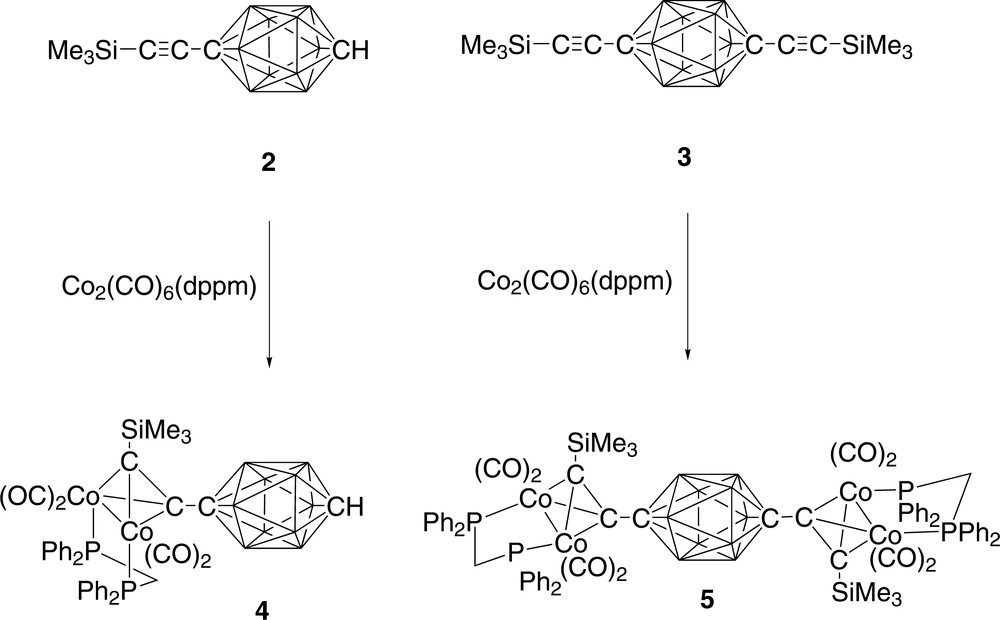
The synthesis of compounds 4 (left) and 5 (right).
The reaction of the diethynyl carborane 3 [13] with two equivalents of Co2(CO)6(μ-dppm) afforded the tricluster 5 in moderate yield (40%). Coordination of each ethynyl moiety in 3 by the Co2(CO)4(μ-dppm) fragment was clearly evidenced from the IR spectrum which contained both ν(BH) and ν(CO) stretches, but no ν(C≡C) band, the characteristic 1H resonances in the NMR spectrum of the dppm ligand methylene protons and a single resonance from the protons of the SiMe3 groups in the correct ratio for 5, and the FAB mass spectrum, which contained a series of fragment ions derived from 5 by loss of CO ligands.
For purposes of comparison, the complexes {Co2(CO)4(μ-dppm)}2(μ-Me3SiC2C6H4C2SiMe3) (6) [25] and {Co2(CO)4(μ-dppm)}2(μ-Me3SiC2C≡CC≡CC2SiMe3) (7) [26] were also obtained, using minor variations of the methods described previously by others.
2.2 Molecular structures
Single crystals of 4 (Fig. 1), 5 [21] and 6 (Fig. 2) suitable for X-ray diffraction were grown by slow diffusion of MeOH into a concentrated CH2Cl2 solution of each complex (Table 1). Significant bond lengths and angles are summarised in either the appropriate figure captions or Table 2, which also includes the appropriate parameters associated with 7 for comparison [26]. All three molecules sit in a symmetry element. Molecule 4 is bisected by a plane parallel to the longest molecular axis and located at (0 1/4 0), and molecules 5 and 6 lay in an inversion centre located at (1/2 0 1/2) and (1/4 1/4 1/4), respectively. The crystal packing is driven by the close-packing principle [27]. Molecules 5 and 6 present small cavities between molecules, where solvent molecules are trapped. The bulkier nature of 5 and 6, containing two dppm moieties, compared to only one dppm moiety in 4, is likely the cause of the reduced packing efficiency of molecules 5 and 6.
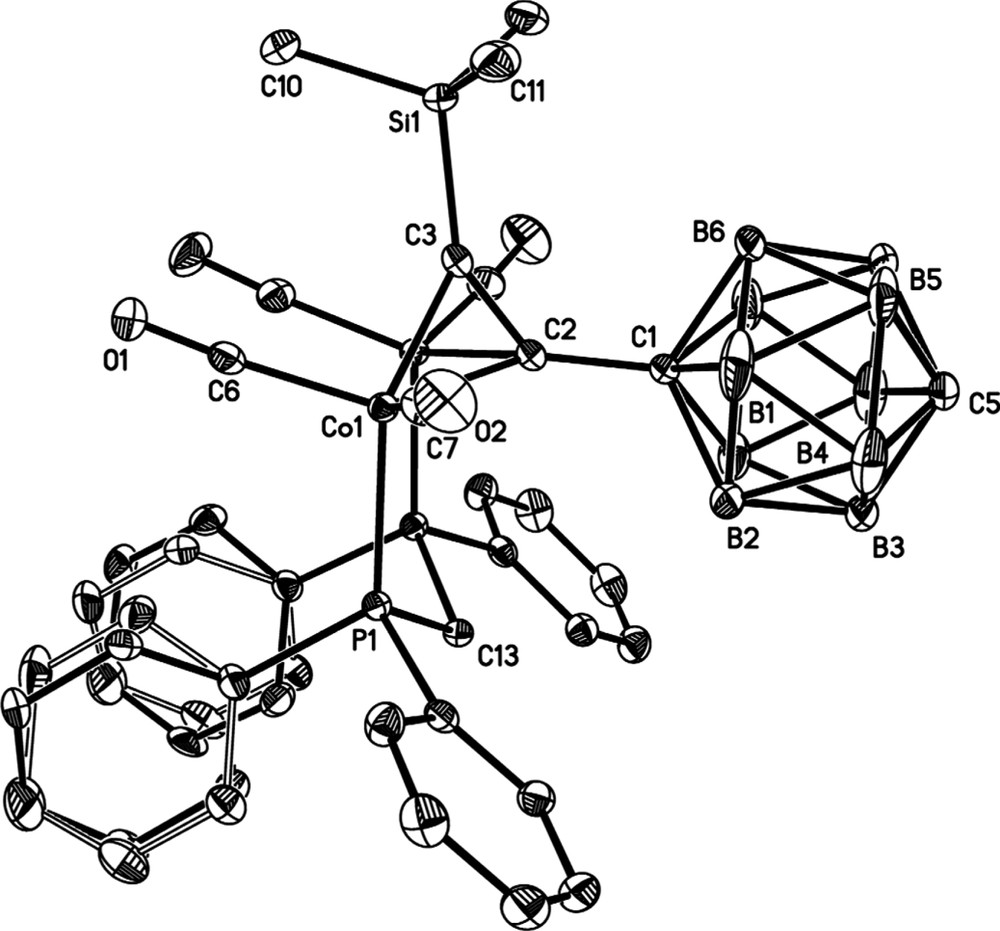
Molecular structure of 4, with hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): C(1)–C(2) 1.510(3), C(2)–C(3) 1.360(3), C(3)–Si(1) 1.852(2), C(1)–B(avg) 1.724, C(5)–B (avg) 1.694, C(1)···C(5) 3.119, Co(1)–Co(1′) 2.4737(4), Co(1)–C(2) 1.977(2), Co(1)–C(3) 1.981(2), Co(1)–P(1) 2.2366(5), C(1)–C(2)–C(3) 132.5(2), C(2)–C(3)–Si(1) 147.6(2).
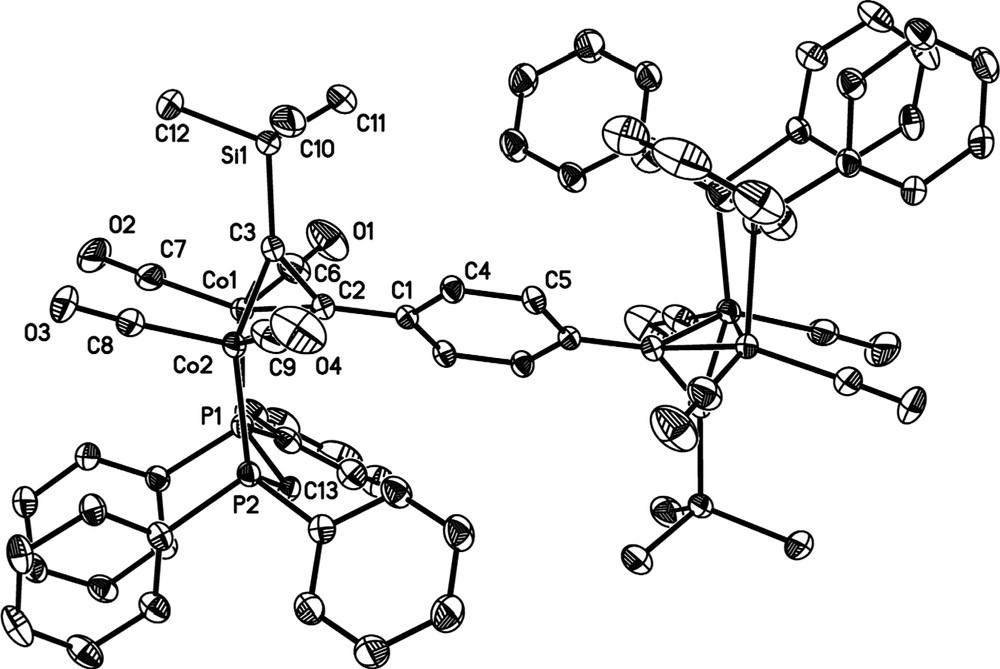
Molecular structure of 6, with hydrogen atoms omitted for clarity.
Crystallographic details for compounds 4–6
| Compound | 4 | 5 | 6 |
| Empirical formula | C36H42B10Co2O4P2Si | C73H78B10Cl6Co4O8P4Si2 | C75H66Cl3Co4O8P4Si2 |
| Formula weight | 854.69 | 1819.93 | 1617.41 |
| Temperature (K) | 100 (2) | 100(2) | 120(2) |
| Crystal system | Orthorhombic | Triclinic | Monoclinic |
| Space group | Pnma | I2/a | |
| a (Å) | 17.3523 (11) | 12.287 (2) | 25.1439 (18) |
| b (Å) | 14.6814 (8) | 13.004 (2) | 12.2653 (9) |
| c (Å) | 15.8841 (9) | 13.545 (2) | 24.2413 (16) |
| α (°) | 90 | 103.148 (4) | 90 |
| β (°) | 90 | 95.820 (4) | 97.313 (2) |
| γ (°) | 90 | 96.040 (4) | 90 |
| Volume (Å3) | 4046.6 (4) | 2078.1 (6) | 7415.1 (9) |
| Z | 4 | 1 | 4 |
| Dc (Mg m–3) | 1.405 | 1.454 | 1.449 |
| μ (mm–1) | 0.967 | 1.135 | 1.160 |
| Crystal size (mm) | 0.40 × 0.25 × 0.20 | 0.32 × 0.20 × 0.08 | 0.38 × 0.24 × 0.20 |
| Theta range (°) | 1.74–27.49 | 1.56 to 27.71 | 1.63–30.52 |
| Reflections collected | 43,663 | 23 469 | 55 546 |
| Independent reflections | 4823 | 9578 | 11310 |
| R(int) | 0.0555 | 0.0396 | 0.0274 |
| wR(F2) (all data) | 0.0731 | 0.0789 | 0.0856 |
| R(F) (all data) | 0.0395 | 0.0387 | 0.0427 |
| Refined parameters | 320 | 507 | 455 |
| Goodness-of-fit | 1.056 | 1.066 | 1.012 |
| Δρmin,max (e Å–3) | 0.400 and –0.420 | 0.732 and –0.661 | 0.427 and –0.516 |
Crystallographic data (distances in Å, angles in °) for compounds 5–7 and principal structural characteristics, relative energies (Erel, kcal mol–1), and adiabatic ionisation potentials (IP, eV) for computational models [5-H]n+– [7-H]n+ (n = 0–2)
| Compound | C(3)–C(2) | C(2)–C(1) | Co–Co | C(3)–Co | C(2)–Co | C(1)–X a,b | X–X a,b | C(3)–C(2)–C(1) | Erel | IP |
| 5 | 1.359 (2) | 1.497 (2) | 2.4804 (4) | 1.728 (3) | 1.772 (4) | 139.3 (2) | ||||
| 5-H | 1.355 | 1.491 | 2.527 |
|
| 1.728 | 139.2 | 6.64 | ||
| [5-H(FS)]+ g | 1.340 | 1.483 | 2.470 |
|
| 1.727 |
| 141.5 | 0.00 | |
| [5-H(BS)]+ h |
|
|
|
|
|
|
|
| 4.23 | |
| [5-H(S)]2+ i | 1.327 | 1.480 | 2.421 |
|
| 1.728 |
| 144.1 | 5.63 | |
| [5-H(T)]2+ j | 1.323 | 1.487 | 2.411 |
|
| 1.726 |
| 143.8 | 0.00 | |
| 6 | 1.357 (2) | 1.467 (2) | 2.4875 (3) |
|
| 1.405 (2) 1.398 (2) | 1.389 (2) | 136.3 (1) | ||
| 6-H | 1.348 | 1.452 | 2.526 |
|
| 1.416 | 1.389 | 142.8 | 6.31 | |
| [6-H]+ | 1.340 | 1.439 | 2.485 |
|
| 1.421 | 1.384 | 146.0 | ||
| [6-H(S)]2+ i | 1.335 | 1.430 | 2.446 |
|
| 1.427 | 1.378 | 149.3 | 0.69 | |
| [6-H(T)]2+ j | 1.329 | 1.448 | 2.420 |
|
| 1.417 | 1.387 | 147.9 | 0.00 | |
| 7 | 1.343 (11) | 1.386 (9) | 2.478 (3) |
|
| 1.210 (10) | 1.372 (14) | 144.4 (9) | ||
| 7-H | 1.363 | 1.368 | 2.530 |
|
| 1.244 | 1.339 | 145.8 | 6.33 | |
| [7-H]+ | 1.353 | 1.352 | 2.494 |
|
| 1.250 | 1.328 | 150.7 | ||
| [7-H(S)]2+ i | 1.347 | 1.337 | 2.455 |
|
| 1.258 | 1.313 | 160.0 | 0.00 | |
| [7-H(T)]2+ j | 1.361 | 1.317 | 2.550 |
|
| 1.269 | 1.295 | 161.5 | 2.62 |
a X = B for 5 and [5-H]n+, X = C(4), C(5) for 6, [6-H]n+, 7 and [7-H]n+.
b Average distances.
c Distance with respect to Co(1).
d Distances with respect to Co(2).
e Average C(1)–B distances.
f Average intra-cluster B–B distance.
g Full symmetry.
h Broken symmetry.
i Low-spin configuration.
j High-spin configuration.
The bis(phosphine) ligand in 4 lies approximately cis to the C2B10H11 icosahedron. The two ordered phenyl rings of the dppm ligand adopt an unusual co-planar arrangement, and this probably reflects the most efficient packing motif within the solid-state structure. The two Co2C2 tetrahedra of 5 are related by a crystallographic inversion centre at the core of the C2B10 icosahedron, and, in contrast to 4, the dppm ligands in 5 are found occupying positions approximately trans to the carborane cage. In the case of the phenyl-substituted derivative the dppm ligands adopt an anti arrangement.
Within the carborane cage portion of 4, the C(5)−B bonds are somewhat shorter [1.687(3)–1.703(3) Å] than for C(1)−B [1.714(3)–1.738(4) Å] while the B−B distances are experimentally equivalent [1.755(4)–1.776(4) Å]. The lengthening of C−B bonds in carborane cages caused by C-substitution is well-documented, although variation in B−B distances is relatively rare [13]. The C(1)−C(2) bond distance [1.510(3) Å] is considerably elongated in comparison to C(carborane)−C(ethynyl) distances in 3 [1.453(2) Å] and diethynyl carborane [1.449(2)/1.452(2) (two independent molecules)], but comparable with that found in 5 [1.497(2) Å]. The C(2)−C(3) [1.360(3) Å], C(2,3)−Co [1.977(2), 1.981(2) Å] and Co–Co [2.4737(4) Å] separations are within the normal limits [28].
The carborane cage in 5 is one of the most spherical structurally characterised examples of a para-carborane known to date. For example, the C(1)···C(1) separation [3.176(7) Å] and average tropical B−B bond lengths [1.772(4) Å] are in sharp contrast with the more ovoid shape of the carborane cages in 3 [12] and 1,12-bis(ethynyl)-para-carborane 8 [C…C 3.104(2) Å; avg. tropical B−B 1.793(3) Å] [13]. The elongation of the C(1)−C(2) and C(2)−C(3) bond lengths in 5 compared to 3 are as expected, and reflect the different nature of the C(2) and C(3) centres in each complex. The parameters associated with Co2C2 clusters fall within the normal ranges.
The molecular parameters of the C6H4 spaced species 6 are comparable with those of other dicobalt complexes of this type derived from donor-substituted phenyl acetylenes [28]: the C(3)−C(2)−C(1)−C(4) torsion angle is 44.7(3)° and there is a small twist of the ligating groups around the Co(1)−Co(2) bond axis away from an ideal eclipsed geometry. The Co(1)−Co(2) bond length [2.488(1) Å] is similar to that observed for Co2(μ-HC2Ph)(CO)4(μ-dppm), while the C(3)−C(2) bond length [1.357(2) Å] is at the longer end of the range usually encountered.
2.3 Electrochemical characterisation
Electrochemical studies have been used to demonstrate communication between two Co2C2 clusters of this type via aryl, alkynyl or ferrocenyl entities [18,29,30]. An examination of the electrochemical response of 4 using cyclic voltammetry revealed both a single oxidation process and a single reduction (E1/2 = +0.82 V, –1.60 V, vs. SCE, decamethylferrocene/decamethylferrocinium (Fc*/Fc*+) = 0.084 V in THF containing 0.1 M [NBu4]PF6). These redox processes were chemically and electrochemically reversible at a moderate scan rate (0.100 V s–1), with ipa/ipc ca. 1 and ΔEp ca. 60 mV, identical to that of the Fc*/Fc*+ couple used as an internal standard. The reversibility of the redox processes is notable, with both the dppm ligand and the steric bulk of the carborane cage likely to be playing a role in the stabilisation of both the oxidised and reduced forms of 4.
A detailed electrochemical study of 5 revealed both oxidation and reduction processes at + 0.87, + 0.77 and –1.58, –1.66 V (vs. SCE, Fc*/Fc*+ = 0.084 V); ΔE0(ox) ca. 105 mV, KC = 60; ΔE0(red) ca. 80 mV, KC = 5], respectively [21]. No redox processes were found within the accessible potential window when 3 was subjected to a similar electrochemical study. For comparison, the phenyl-bridged complex [{Co2C2(H)(CO)4(μ-dppm)}2(μ-1,4-C6H4)] (6) also gave two oxidation and reduction processes with ΔE0(ox) 110 mV, ΔE0(red) 80 mV. The electrochemical response of the butadiyndiyl spaced derivative [{Co2C2(SiMe3)(CO)4(μ-dppm)}2(μ-C4)] (7) was complicated by the chemical irreversibility of the redox processes. Two chemically irreversible oxidations were observed with peak potentials +0.99 and +1.26 V, and single reduction at –1.32 V. On the basis of this electrochemical work it is not possible to identify or quantify the electronic interactions occurring between the Co2C2 redox centres via the C4 bridge.
The difference in oxidation potentials observed in 5 and 6 is significant, especially given the > 6 Å separation of the cobalt cluster cores, and indicates a moderate degree of electronic communication between the cobalt clusters in the radical cations 5+ and 6+. The separation of the reduction events is somewhat smaller, but is still larger than the statistical limit (ca. 36 mV at 298 K) predicted for non-interacting redox centres [30]. The electrochemical studies clearly indicate that the bonding framework of the para-carborane cage and the para-C6H4 ring provides a conduit for electronic effects between the cluster substituents in the 1,12 and 1,4 positions, respectively, as a through space mechanism would perhaps be expected to give rise to more similarly spaced oxidation and reduction processes, although such process could be influenced by solvation and charge effects.
In general terms, the interaction between the orbitals of a M2L6 fragment with those of an acetylenic fragment gives rise to a pronounced bending of the acetylenic chain and to a splitting of both the π and π* C–C orbitals [31]. If these orbitals are located in the HOMO–LUMO region, or mix with the frontier orbitals of a spacer group, they will govern the chemistry of this type of complex, and in this context it is important to note that the Co2C2 tetrahedron offers a high-lying fragment orbital of the correct symmetry to interact with aryl [28] and ethynyl [19c] groups. In light of the electrochemical properties of 5, 6 described above and elsewhere, and the ongoing interest in redox-active organometallic species spanned by C4 [32], it was of interest to probe the mechanism through which electronic effects are transmitted between Co2C2 fragments via the 1,12-para-carborane cage, the 1,4-C6H4 ring and the butadiyndiyl fragment in more detail.
2.4 Theoretical description
Density functional-theory (DFT) molecular-orbital calculations were carried out on dicobalt complexes of 1,12-diethynyl-para-carborane (5-H), 1,4-diethynylbenzene (6-H) and 1,3,5,7-octatetrayne (7-H) in order to further the understanding of their electronic properties1. In order to verify the accuracy of the computational method, ‘free’ 1,12-diethynyl-para-carborane of D5d symmetry (3-H) was studied in the first instance, although we note that the electronic structure of 3-H has been calculated at the ab initio Hartree–Fock (HF) level of theory on a previous occasion [12]. Pertinent results from the geometry optimisations are shown in Table 3, together with the experimentally observed values. Both HF and DFT optimised structures of 3-H compare well with the crystallographically determined structure of 8 [13]. Interestingly enough, the C≡C distances are slightly better reproduced at the HF level since the largest deviation is only 0.005 Å, whereas a deviation of 0.030 Å is observed at the DFT level. On the other hand the DFT computed B–B and B–C distances of the carborane cluster are in a better agreement with the X-ray structure. The maximum deviation concerns some B–B distances computed 0.007 Å shorter than the experimental values.
Principal structural characteristics (distances in Å, angles in °) for the experimental structure 3 and the computational model 3-H
| 8[13] | 3-H | ||
| HF[12] a | DFT | ||
| C(3)–C(2) | 1.180 (3) | 1.185 | 1.210 |
| C(2)–C(1) | 1.451 (2) | 1.449 | 1.440 |
| C(1)–B(avg) | 1.726 (3) | 1.796 | 1.786 |
| B–B(avg) b | 1.793 (3) | 1.723 | 1.726 |
| B–B(avg) c | 1.761 (3) | 1.769 | 1.760 |
| C(1)–C(2)–C(3) | 179.18 | 180.0 | 180.0 |
| C(2)–C(1)–B | 117.91 | 117.6 | 118.3 |
a HF/6-31G*.
b Distances between atoms within a B5 ring.
c Intra-cluster B–B distances.
The hydrogen-substituted model complex [Co2(CO)4(PH3)2]2(μ-H3SiC2CB10H10CC2SiH3) (5-H) of Ci symmetry, was used to mimic complex 5. Model 5-H quite satisfactorily mimics the metric distances of the crystallographically determined structure (Table 2). The computed Co–Co distance is slightly overestimated (2.527 vs. 2.4804(4) Å), mainly due to the use of the Becke–Perdew non-local corrections. The important geometry variations between 3 and 5 noted experimentally are reproduced in the computed results and, for example, coordination of the Co2 groups results in a large increase in the ethynyl C–C bond length and a smaller increase in the C(sp)–C(sp3) separation. The calculated B–B distances are virtually unaffected by coordination of the ethynyl moieties to the metal centres.
2.5 Electronic structure
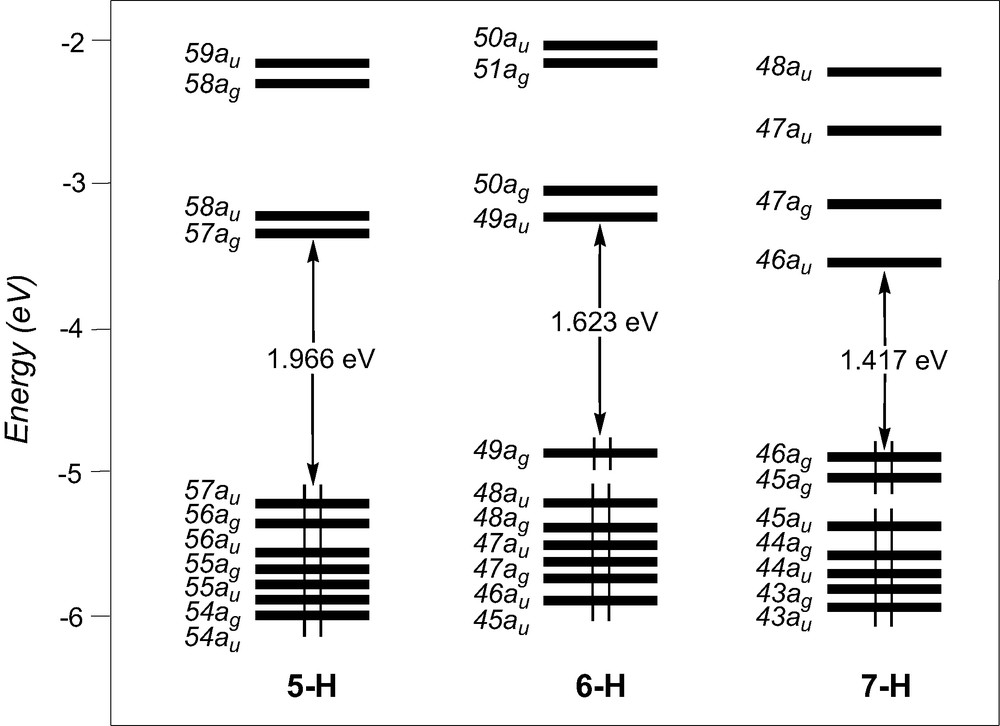
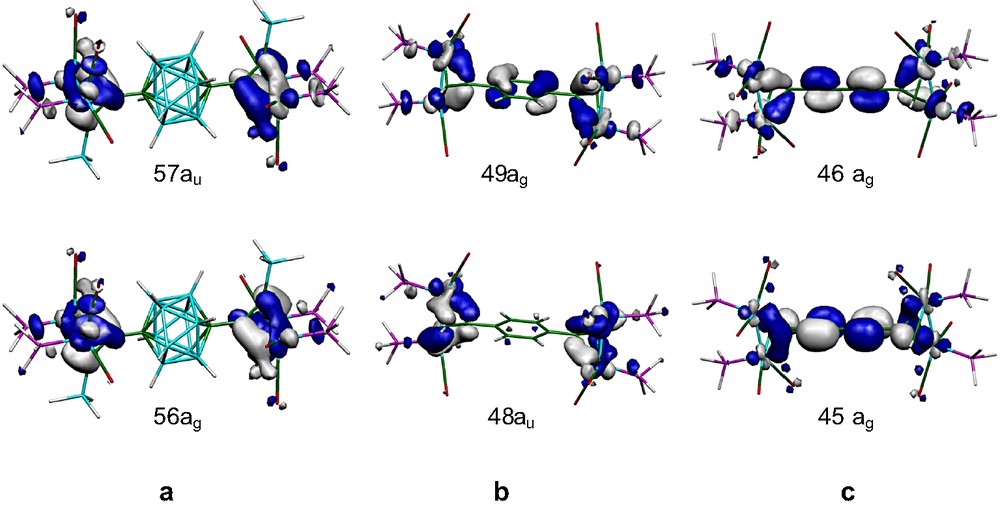
The DF molecular orbital diagram of the neutral complex 5-H is shown on the left-hand side of the Fig. 3. A large energy gap (1.966 eV) separates the HOMOs and a set of two LUMOs (57ag and 58ag) which are also separated from the other vacant orbitals by a gap of around 1 eV. It is informative to look at the composition of the HOMOs of 5-H before considering the structural and electronic changes induced by oxidation. To this end a Mulliken atomic orbital population analyses for selected MOs of 5-H is given in the Table 4. It appears that the orbitals of the HOMO–LUMO region present a preponderant metallic character. For instance, the HOMOs 57au and 56ag are 71% and 73% Co in character, respectively, with a weak participation of the carbon atoms C(3) and C(2). The rest of the electronic contribution is localised on the PH3 and CO ligands bound to the metallic atoms. These MOs are mainly of π-type Co–Co antibonding. These results are displayed graphically in Figs. 3 and 4a.
DFT molecular orbital diagrams of 5-H (left), 6-H (middle), and 7-H (right).
Energy (ε, eV), occupation and localisation (%) of the frontier molecular orbitals in the neutral complexes 5-H, 6-H and 7-H
| 5-H | 58au | 57ag | 57au | 56ag | MO | 55ag | 55au | 54ag |
| ε | –3.25 | –3.30 | –5.27 | –5.36 | –5.60 | –5.65 | –5.76 | –5.80 |
| Occ | 0 | 0 | 2 | 2 | 2 | 2 | 2 | 2 |
| C(1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| C(2) | 7 | 7 | 9 | 8 | 4 | 2 | 3 | 2 |
| C(3) | 7 | 5 | 5 | 8 | 6 | 6 | 8 | 6 |
| Co | 70 | 70 | 71 | 73 | 77 | 80 | 80 | 80 |
| PH3 | 2 | 3 | 10 | 7 | 2 | 2 | 2 | 3 |
| CO | 14 | 15 | 5 | 4 | 11 | 10 | 7 | 9 |
| B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6-H | 50ag | 49au | 49ag | 48au | 48ag | 47au | 47ag | 46au |
| ε | –3.01 | –3.25 | –4.87 | –5.23 | –5.45 | –5.50 | –5.58 | –5.67 |
| Occ | 0 | 0 | 2 | 2 | 2 | 2 | 2 | 2 |
| C(1) | 1 | 5 | 12 | 0 | 2 | 0 | 2 | 0 |
| C(2) | 8 | 11 | 5 | 7 | 0 | 4 | 0 | 4 |
| C(3) | 8 | 2 | 0 | 9 | 2 | 5 | 12 | 3 |
| C(4) | 1 | 6 | 8 | 2 | 0 | 0 | 0 | 0 |
| Co | 67 | 64 | 61 | 72 | 83 | 78 | 78 | 82 |
| PH3 | 0 | 2 | 10 | 5 | 0 | 2 | 1 | 2 |
| CO | 15 | 10 | 4 | 5 | 13 | 11 | 7 | 9 |
| 7-H | 47ag | 46au | 46ag | 45ag | 45au | 44ag | 44au | 43ag |
| ε | –3.16 | –3.52 | –4.94 | –5.06 | –5.44 | –5.64 | –5.69 | –5.80 |
| Occ | 0 | 0 | 2 | 2 | 2 | 2 | 2 | 2 |
| C(1) | 2 | 7 | 15 | 32 | 0 | 2 | 0 | 0 |
| C(2) | 8 | 10 | 4 | 7 | 7 | 0 | 2 | 0 |
| C(3) | 7 | 0 | 0 | 11 | 8 | 3 | 5 | 11 |
| C(4) | 4 | 8 | 11 | 22 | 5 | 1 | 1 | 0 |
| Co | 64 | 61 | 56 | 25 | 71 | 78 | 80 | 80 |
| PH3 | 0 | 4 | 10 | 3 | 5 | 2 | 2 | 2 |
| CO | 15 | 10 | 4 | 0 | 4 | 14 | 10 | 7 |
Molecular orbital plots for the HOMO (top) and HOMO-1 (bottom) of 5-H (a), 6-H (b), and 7-H (c). Contour values are ±0.03 (e/bohr3)1/2.
Using a Ci restrained geometry, one electron was removed from the structure 5-H and the structure re-optimised. For comparison, calculations were also carried on the broken symmetry geometry [5-H]+ (C1 symmetry). A comparison of the structural parameters calculated for 5-H and [5-H]+, both with full symmetry (FS) and broken symmetry (BS) reveals in a decrease in the metal-metal bond length, a contraction of C(3)–C(2) bond and a lengthening of the various C–Co separations upon oxidation. The weak antibonding and bonding character of the HOMO with respect to these connections provides a simple rationalisation of these structural variations. Interestingly, a slight asymmetry is computed for [5-H(BS)]+ (see Table 2), which is less stable than [5-H(FS)]+ by 4.23 kcal mol–1. The latter result suggests than such a geometry, which would favour some valence-localised structure character, is unlikely with respect to the valence-delocalised structure.
A study of the spin density of the complex [5-H]+ indicates a net localisation of the single electron on the Co atoms with some uneven distribution over Co1 and Co2 (see Table 5). The DFT calculations indicate that the π-orbitals on the carborane entity do not participate much in the partially occupied MO and therefore should not play an important role in the communication between the metallic clusters in the complex 5. Rather, the separation of the electrochemical events observed voltammetrically must be attributed to inductive effects transmitted by polarisation of the σ-bonding framework.
Calculated distribution of spin density in the cationic open-shell complexes [5-H]n+–[7-H]n+ (n = 1, 2)
| Complex | Co(1) | Co(2) | C(3) | C(2) | C(1) | Si | P | B a | C(4) |
| [5-H(FS)]+ | 0.239 | 0.187 | –0.009 | 0.018 | –0.005 | –0.004 | 0.022 | 0.006 | |
| 0.034 | |||||||||
| [5-H(BS)]+ | 0.312 | 0.153 | –0.015 | 0.026 | –0.006 | –0.003 | 0.050/0.023 | 0.008 | |
| 0.208 | 0.145 | –0.007 | 0.018 | –0.004 | –0.003 | 0.040/0.014 | 0.007 | ||
| [6-H]+ | 0.228 | 0.175 | –0.027 | –0.005 | 0.045 | 0.035 | 0.019 | ||
| 0.030 | –0.001 b | ||||||||
| [7-H]+ | 0.227 | 0.166 | –0.030 | –0.008 | 0.048 | 0.034 | 0.027 | 0.027 | |
| 0.032 | |||||||||
| [5-H(T)]2+ | 0.528 | 0.317 | –0.015 | 0.035 | –0.009 | –0.007 | 0.041 | 0.009 | |
| 0.073 | |||||||||
| [6-H(T)]2+ | 0.574 | 0.256 | –0.024 | 0.020 | 0.023 | 0.074 | 0.017 | ||
| 0.036 | 0.010 b | ||||||||
| [7-H(T)]2+ | 0.319 | 0.270 | –0.017 | 0.040 | 0.142 | 0.059 | 0.095 | ||
| 0.058 |
a Sum over the non-equivalent boron atoms.
b C(4′).
Attention is next turned to the structures and spin states of the dicationic species [5-H]2+. Oxidation of 5-H to the high-spin (triplet) state [5-H(T)]2+, which was calculated to be some 5.63 kcal mol–1 more stable than the low-spin (singlet) state [5-H(S)]2+, does not result in any notable structural modification of the carborane moiety or the C(2)–C(1) distance. However, the intra-cluster Co2C2 bond distances are more significantly affected. The metal–metal distance decreases by some 0.116 Å (4.5%), while there is a smaller contraction of the C(3)–C(2) bond length (2.5%). The C–Co distances increase between 3% and 4% overall. Again, these calculated results are consistent with the nodal properties of the HOMOs (vide supra) and with an interpretation of the electrochemical response in terms of Co2C2 centred oxidation processes.
2.6 Influence of the nature of the spacer entity
In order to compare the electronic properties of the complex 5 with those of the complexes [Co2C2(SiMe3)(CO)4(μ-dppm)]2(μ-1,4-C6H4) (6) and [Co2C2(SiMe3)(CO)4(μ-dppm)]2(μ-C≡CC≡C) (7), DFT calculations have been carried out on the model complexes [Co2C2(H)(CO)4(μ-dppm)]2(μ-1,4-C6H4) ([6-H]n+) and [Co2C2(H)(CO)4(μ-dppm)]2(μ-C≡CC≡C) ([7-H]n+) (n = 0–2). The optimised structural characteristics of these two model complexes are given in Table 2. The optimised distances of the neutral complexes 6-H and 7-H are in good agreement with the crystallographically determined structures, with some over-estimation of the Co–Co distances attributed to the previously mentioned methodological reasons. Both the observed and calculated C(3)–C(2)–C(1) angles vary across the series of complexes 5 (5-H), 6 (6-H) and 7 (7-H) being 139.2(2) (139.2°), 136.5(3) (145.8°) and 144.4(9) (142.8°), respectively (see Figs. 1 and 2 for a representative atom numbering scheme). As might be expected, the bulkier the spacer entity, the more pronounced the bending of the chain, although the observed deviation from linearity is greater in the case of 6 than 6-H.
The DF molecular orbital diagrams for the neutral complexes 6-H and 7-H are shown in Fig. 3, and illustrate the large HOMO–LUMO gap calculated in each case (1.623 and 1.417 eV, respectively). The HOMO and HOMO-1 of the complexes 6-H and 7-H are shown graphically in Fig. 4, with the energy and composition of these orbitals given in Table 5. The HOMO 49ag and HOMO-1 48au of the neutral complex 6-H are mainly localised on the Co atoms (61% and 72%, respectively). The HOMO also contains a small contribution from the benzene spacer (12% C(1) and 8% C(4)) which is not present in the HOMO-1. In much the same way as has been described for 5-H, the strong antibonding character of the Co–Co and C(2)–C(3) interactions in these frontier orbitals helps explain the reduction of the Co–Co bond length calculated to occur upon oxidation, while the bonding nature of the Co–C (2, 3) interaction is consistent with the elongation of these bonds (Table 2). The elongation of the C(1)–C(4) bond and contraction of C(4)–C(4′) give some indication of a quinodal form, and suggests a more intimate role of the 1,4-C6H4 π-system in the interactions between the cluster centres than was found for the carborane cage.
The oxidation of 6-H to [6-H]2+ leads to two spin states (HS and LS), which are isoenergetic (less than 1 kcal mol–1 in favour of the triplet state). As would be expected on the basis of the orbital composition of the HOMO and HOMO-1 in 6-H, in the case of the LS state the structural modifications take place on the whole Co2C2–C6H4–C2Co2 chain, and lead to a further emphasis of the quinoidal character of the bridge. In the case of the HS state, the greatest structural changes of significance are restricted to the intracluster Co2C2 distances (Table 2).
The HOMO (46ag) and HOMO-1 (45ag) of the complex 7-H, which lie close together in energy, are slightly separated from the other occupied orbitals, are of π-type and mainly localised on the Co atoms and on the C atoms of the spacing group. Whereas the HOMO is rather localised on the Co atoms, with a small, but significant contribution from the C4 bridge (56% Co against 4%, 15% and 11% for the C(2), C(1) and C(4) atoms), the HOMO-1 presents an even more delocalised character with 25%, 11%, 7%, 32% and 22% on the Co, C(3), C(2), C(1) and C(4) atoms, respectively. Moreover, the bonding-antibonding properties of these orbitals are not totally equivalent and, for example, the Co–Co antibonding character of the HOMO is not observed in the HOMO-1.
In a manner entirely analogous to the monooxidation of 5-H and 6-H, oxidation of 7-H to [7-H]+ results in contraction of the Co–Co, C(1)–C(2) and C(2)–C(3) bonds reflecting the composition of the redox-active orbital. The structural parameters of the C4 bridge are rather less affected than is generally the case of σ-bonded diyndiyl complexes [32], with a small elongation of the formal C(3)≡C(4) triple bond and contraction of the C(4)–C(4′) single bond (Table 4).
The structural parameters of the Co2C2–C4–C2Co2 chain are further affected by oxidation to [7-H]2+ (Table 2). In the LS state of [7-H]2+, which is be calculated to be slightly more stable than the HS state by 2.62 kcal mol–1, the Co–Co, C(3)–C(2), C(2)–C(1) and C(4)–C(4') distances decrease by 0.075, 0.016, 0.031 and 0.026 Å, respectively, when compared with 7-H, whereas the C(1)–C(4) bond length increases by 0.014 Å. The intra-cluster Co–C and the intra-bridge C(1)–C(4) bond lengths are further increased by this second oxidation process. Overall, the structural and electronic parameters of the oxidised forms of 7-H support a significant contribution from the C4 π-system in promoting delocalisation of electron density between the Co2C2 centres, and an evolution from a formally diyndiyl C(1)≡C(4)–C≡C description to a more cumulenic C(1)=C(4)=C=C form. This is further supported by the increasing linearity of the C(3)–C(2)–C(1) fragment as oxidation proceeds.
The atomic spin densities calculated for [6-H]+ and [7-H]+ (Table 5) show that the unpaired electron is also mainly located on the Co atoms, as noted previously for [5-H]+. Nevertheless, the amount of spin density localised on the metal centres drops from 85% for [5-H]+ to 80% and 78% for [6-H]+ and [7-H]+ with an increasing amount of the unpaired electron spin found on the organic bridge, indicating a tendency for a somewhat higher delocalisation for the latter. The trend is even higher in the case of the dicationic high-spin species, in particular with complex 7-H, since 84%, 83%, and 59% of the two unpaired electrons are located at the cobalt centres for [5-H]+, [6-H]+, and [7-H]+, respectively.
2.7 Ionisation potentials
The first and second ionisation potentials have been computed for the three complexes 5-H (6.64 and 9.04 eV), 6-H (6.31 and 9.08 eV) and 7-H (6.33 and 9.14 eV) and are not strongly influenced by the nature of the bridging group, strongly supporting the concept that the oxidation processes will mainly affect the common part of these molecules i.e. the Co2C2 cluster. However, the larger difference between the first and second IPs for the complexes 6-H and 7-H with respect to 5-H (2.77, 2.81 vs. 2.40 eV), which is in agreement with the experimental electrochemical results from the real systems, gives some evidence for greater thermodynamic stability of the mono-oxidised forms [6-H]+ and [7-H]+, which may be due in part to the better electronic interaction between the Co2C2 clusters when linked by the aryl or ynyl moieties than the carborane.
3 Conclusion
On the basis of electrochemical measurements, the degree of electronic ‘communication’ between cluster-based redox probes Co2C2(CO)4(μ-dppm) through 1,12-C2B10H10 and 1,4-C6H4 moieties is comparable. Examination of the electronic structures of these systems, together with the related C4 spaced analogue by DFT methods reveals a different mechanism for the passage of these interactions is in play in each case. The carborane cluster cage acts as a more or less purely σ-based bridging entity: a mismatch between orbitals of the carborane and the pi-orbitals of the ethynyl group is the main reason for the absence of carborane participation in the frontier orbitals of this compound. Crucially, the electrochemical events observed do not relate to the ‘aromaticity’ of the bridge and MOs with strongly delocalised on the carborane may be found at lower energy. The role of the bridge π-orbitals is more significant in the case of 1,4-C6H4 and even more so in C≡CC≡C bridged analogues.
4 Experimental
4.1 General conditions
All reactions were carried out using standard Schlenk techniques under dry high-purity nitrogen. Solvents were dried and distilled prior to use. Preparative TLC was carried out on 20 × 20 cm glass plates coated with silica gel (Merck G254, 0.5 mm thick). Reagents were purchased and used as received. Compounds 3 [13], 6 [25] and 7 [26] were prepared according to the literature methods with minor modifications. Compound 2 was made using a similar procedure to the reported synthesis of 3. IR spectra were recorded on a Nicolet Avatar spectrophotometer using solution cells fitted with CaF2 windows. NMR spectra were obtained from solutions in CDCl3 using Varian VXR-400 (1H 399.97, 13C 100.57, 31P 161.1 MHz) or Bruker DRX-400 (1H 400.13, 13C 100.61, 31P 162.05 MHz) spectrometers. FAB-MS were recorded at the EPSRC National Mass Spectrometry Service Centre (Swansea). Electrochemical experiments were conducted in THF solution containing 0.1 M [NBu4][PF6] using a simple three electrode cell and an EcoChemie Autolab PGSTAT-30, or an EG&G 273 potentiostat/galvanostat. Working electrodes were glassy carbon, with Pt wire counter and pseudo-reference electrodes. Potentials were corrected to SCE using an internal decamethylferrocene/decamethylferrocinium couple as standard (Fc*/Fc*+ = 0.084 V).
4.2 Preparation of [Co2(CO)4(μ-dppm)](μ-Me3SiC2CB10H10CH) (4)
A solution of [Co2(CO)6(μ-dppm)] (100 mg, 0.15 mmol) was treated with 2 (36 mg, 0.15 mmol) and heated at reflux point for 3 h. The solution was cooled, the solvent removed and the residue purified by preparative TLC (70:30 hexane/CH2Cl2). The major band was collected and crystallised (CH2Cl2/MeOH) to afford 4 (50 mg, 40%). ES-MS (m/z) 855, [M]+; 827, [M–CO]+. IR (cyclohexane}: v (BH) 2656; v(CO) 2027 m, 2002 s, 1977 s cm–1. NMR (CDCl3, δ) 1H{11B} 7.38–7.20 (m, 20H, Ph), 3.51, 3.37 (2 × dt, JHP = JHH = 12 Hz, 2 × 1H, CH2), 2.61 (br, 6H, B7-11H + carboranyl CH), 2.15 (br, 5H, BH), 0.27 (s, 9H, SiMe3); 11B –10.8 (5B, B2-6), –15.6 (5B, B7-11); 31P 32.7.
4.3 Preparation of [Co2(CO)4(μ-dppm)]2(μ-Me3SiC2CB10H10CC2SiMe3) (5)
A solution of 3 (83 mg, 0.24 mmol) in benzene (10 ml) was treated with [Co2(CO)6(μ-dppm)] (400 mg, 0.60 mmol) and heated at reflux for 2.5 h. The solvent was removed in vacuo and the dark brown residue obtained was purified by preparative TLC (CH2Cl2/hexane, 3:7) and crystallisation (CH2Cl2/MeOH) (150 mg, 40%). NMR (CDCl3, δ) 1H{11B} 7.10–6.30 (m, 20H, Ph), 3.09, 3.25 (2 × dt, JHP = JHH = 11 Hz, 2 × 1H, CH2), 1.28 (br, 5H, BH), 0.00 (s, 9H, SiMe3); 11B –11.0; FAB-MS (m/z) 1454, 1426, 1342 [M–nCO]+ (n = 4, 5, 7). IR (cyclohexane): ν(BH) 2659; ν(CO) 2026 m, 2002 s, 1976 s, 1964 m cm–1. Found: C, 52.15%; H 4.56%. C70H72B10O8Si2P4Co4.0.5CH2Cl2 requires C, 52.64%; H 4.42%.
5 Theoretical calculations
Density functional theory (DFT) calculations were carried out on model compounds derived from the experimental structure data using the Amsterdam density functional (ADF) program [33] developed by Baerends et al. [34]. Electron correlation was treated within the local density approximation (LDA) in the Vosko–Wilk–Nusair parameterisation [35]. The non-local corrections of Becke and Perdew were added to the exchange and correlation energies, respectively [36,37]. The numerical integration procedure applied for the calculations was developed by te Velde et al. [33]. The atom electronic configurations were described by a triple-ζ Slater-type orbital (STO) basis set for H 1s, B 2s and 2p, C 2s and 2p, Si 3s and 3p, and P 3s and 3p, augmented with a 3d single-ζ polarisation function for B, C, Si, and P atoms and with a 2p single-ζ polarisation for H atoms. A triple-ζ STO basis set was used for Co 3d and 4s, augmented with a single-ζ 4p polarisation function. A frozen-core approximation was used to treat the core shells up to 1s for B and C, 2p for Si and P, and 3p for Co [33]. Full geometry optimisations were carried out using the analytical gradient method implemented by Verluis and Ziegler [38]. Spin-unrestricted calculations were performed for all the open-shell systems. When needed, calculations using the broken-symmetry formalism (employing an asymmetry in the initial spin densities upon the two Co2 groups [39] were carried out to compare valence-delocalised and presumably valence-localised geometries. Representation of the molecular orbitals was done using MOLEKEL4.1 [40].
6 Crystallography
The X-ray data sets for compounds 4, 5 and 6 were collected on Bruker 3-circle diffractometers with SMART 1K (for 4 and 5) or SMART 6K (for 6) CCD area detectors, using graphite-monochromated sealed-tube Mo Kα radiation. The data collection were carried out at 100 K (for 4 and 5) and 120 K (for 6) using cryostream (Oxford cryosystem) open flow N2 cryostats. Reflection intensities were integrated using the SAINT program, version 5.0 [41] (for 4 and 5) and version 6.2a [42] (for 6).
The crystal structures were solved using direct methods and refined by full-matrix least-squares against F2 of all data using SHELXTL software [41]. All non-hydrogen atoms where refined in anisotropic approximation. All phenyl group hydrogen atoms were placed in calculated positions and refined using a riding model. The crystal structures of 5 and 6 present disordered solvent molecules. Thus, three disordered molecules of CH2Cl2 were found per formula unit in the crystal structure of 5 [21], and one disordered molecule of CHCl3 in the crystal structure of 6. Crystal data and experimental details are listed in Table 1. The CCDC deposition numbers for compounds 4–6 are 250,935, 167,400 and 250,936, respectively.
Acknowledgements
We thank Johnson Matthey plc for a generous loan of RuCl3·n H2O, and the EPSRC National Mass Spectrometry Service Centre (Swansea) for FAB-MS measurements. B.L.G., K.C. and J.-F. H. thank the ‘Pôle de calcul intentif de l'Ouest’ (PCIO) of the University of Rennes and the ‘Institut de développement et de ressources en informatique scientifique’ (IDRIS–CNRS) for computing facilities. These studies were facilitated by travel grants from CNRS, France (DRI-12015). We also thank the EPSRC and the University of Durham for financial assistance. MAF held an EPSRC Advanced Fellowship, MAJP held an EPSRC CASE studentship with Avecia Ltd.
1 Throughout this study, the designator ‘-H’ refers to a computational model system, as distinct from the real compound.