

1 Introduction
In light of their unique electronic properties, fullerene derivatives are suitable building blocks for the preparation of molecular devices displaying photoinduced energy and electron transfer processes [1]. Whereas research focused on the use of C60 as the acceptor in covalently bound donor–acceptor pairs has received considerable attention [1], only a few related examples of fullerene-containing non-covalent systems have been described so far [2,3]. The assembly of the two molecular components by using supramolecular interactions rather than covalent chemistry appears however particularly attractive since the range of systems that can be investigated is not severely limited by the synthetic route. As part of this research, we have recently developed a non-covalent approach based on the self-assembly of C60 derivatives bearing an ammonium unit with crown ethers for the preparation of photoactive supramolecular systems. The ammonium–crown ether interaction itself is however weak [4]. Consequently, the binding constants are rather low and only a small fraction of the two components are effectively associated in solution. In this paper, we now summarize our ongoing progresses in the design of supramolecular systems with additional recognition elements in order to increase the binding constants.
2 Supramolecular porphyrin–fullerene conjugates
The photophysical properties of porphyrin–fullerene hybrids have been studied in details and intramolecular processes such as electron and energy transfer evidenced in such multicomponent hybrid systems [5]. In order to understand the nature of the dialog between the C60 and the porphyrin chromophores, the topology of the two moieties in dyads has been systematically varied and a wide range of covalent assemblies have been reported [5]. A few examples of supramolecular C60–porphyrin conjugates have also been described so far [2]. In most cases, these non-covalent assemblies have been obtained from a C60 derivative bearing a pyridyl moiety and a metalloporphyrin through coordination to the metal ion. Due to the apical binding on the porphyrin subunit, the attractive van der Waals interaction of the fullerene sphere with the planar π-surface of the porphyrin seen for several covalent C60–porphyrin derivatives [6] or in the solid state structures of porphyrin/fullerene co-crystals [7] is not possible in such arrays. Interestingly, during the studies of the non-covalent C60–porphyrin ensemble obtained from methanofullerene derivative 1 bearing an ammonium function and porphyrin–crown ether conjugate 2 (Fig. 1), we found that π-stacking interactions between the two chromophores can have a dramatic effect on the recognition interactions [8].
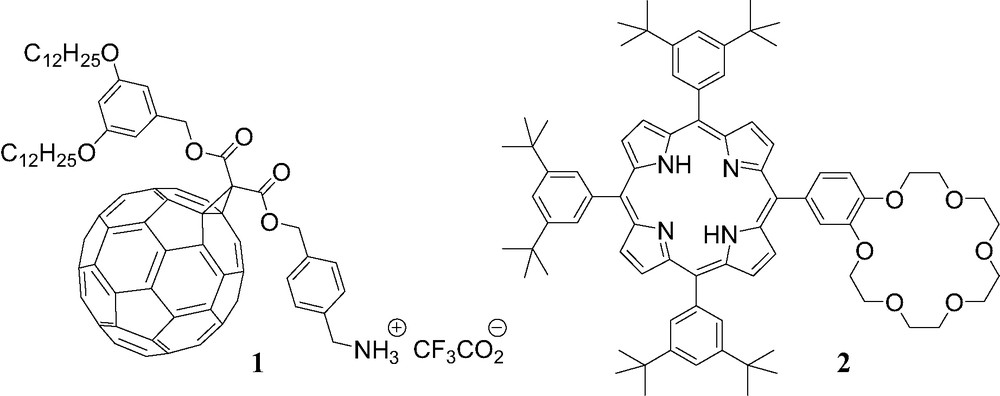
Compounds 1 and 2.
The ability of porphyrin 2 to form a supramolecular complex with ammonium 1 was first evidenced by 1H-NMR binding studies performed at 298 K in CDCl3. As depicted in Fig. 2, the comparison between the 1H-NMR spectra of 1, 2 and mixtures of both components revealed the apparition of new sets of signals as well as complexation-induced changes in chemical shifts. This is particularly visible for the pyrrolic protons of the porphyrin moiety and the signals arising from the crown ether subunit. The latter observations suggest the existence of two conformers for the supramolecular complex obtained upon association of the two components, one being in fast exchange (A) with uncomplexed 1 and 2 on the NMR time-scale, the other one (B) in slow exchange as shown in Fig. 3.
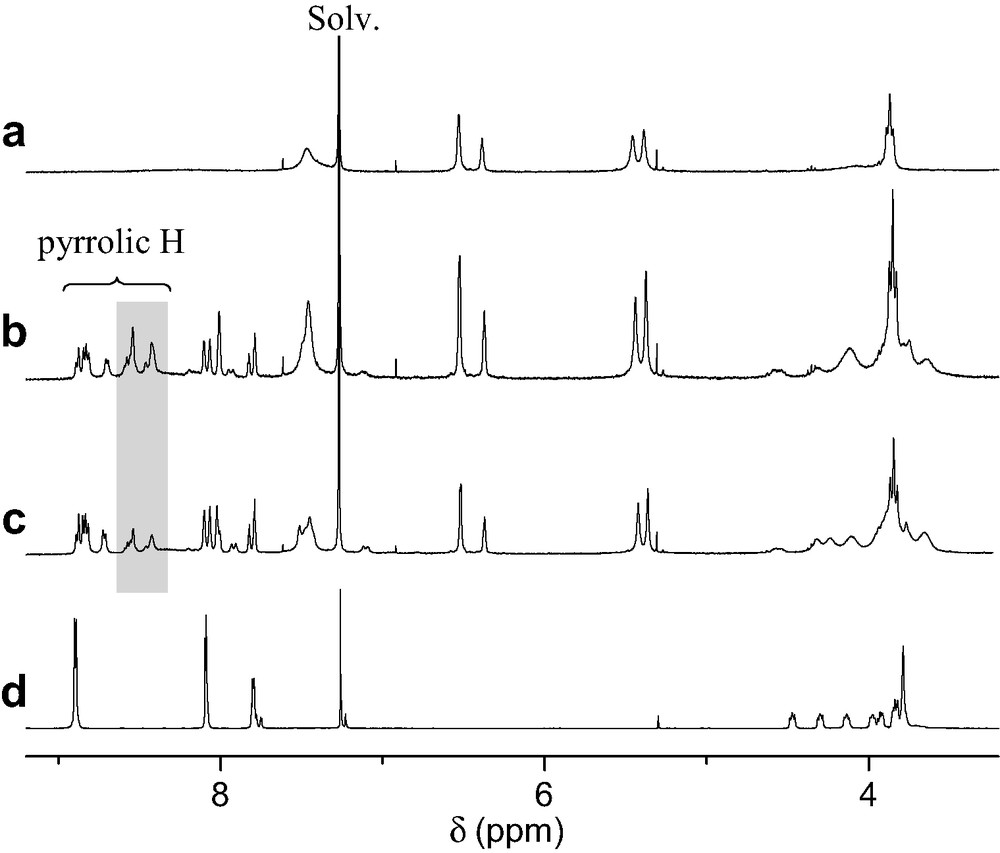
1H-NMR spectra (CDCl3, 300 MHz, 298 K) of 1 (a), 2 (d), a 2:1 mixture of 1 and 2 (b) and a 1:1 mixture of 1 and 2 (c); the signals of the pyrrolic protons of B are highlighted.
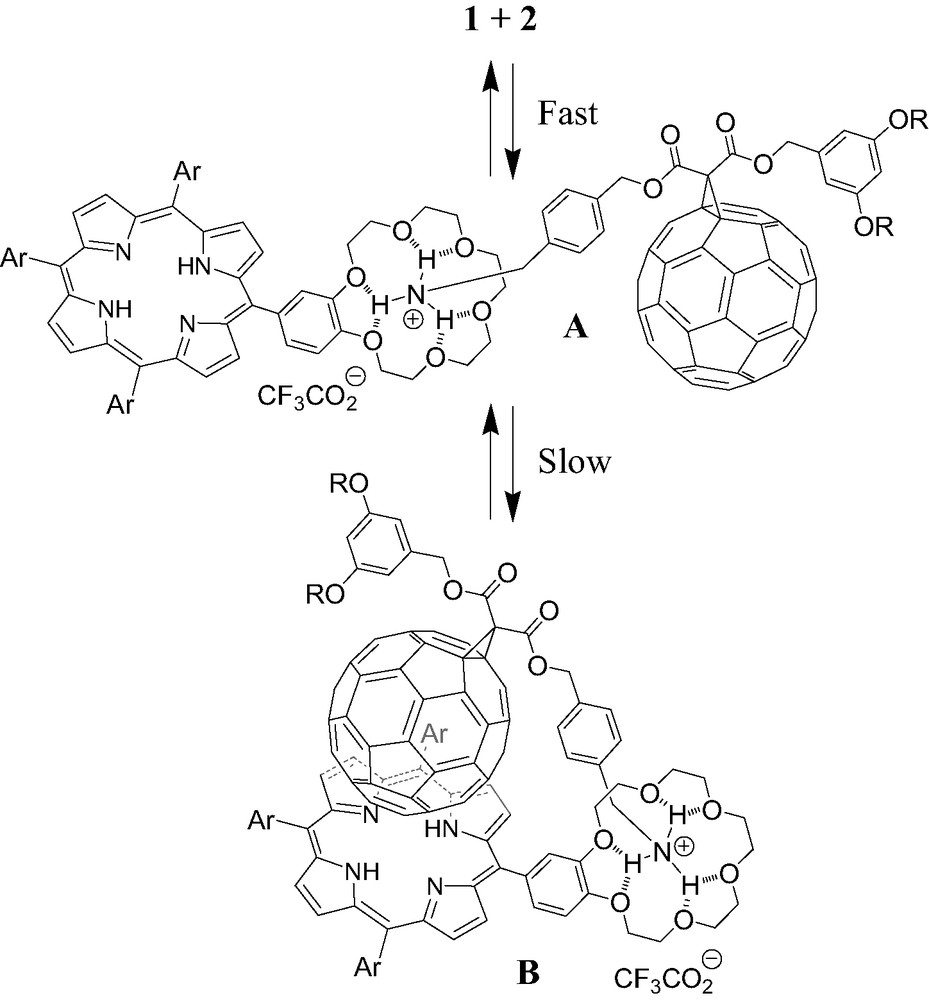
Schematic representation of the two conformers of the supramolecular complex obtained upon association of 1 and 2. A is in fast exchange with uncomplexed 1 and 2 on the NMR time-scale and B in slow exchange.
On the one hand, the initial ammonium–crown ether association leading to conformer A is responsible for the observed complexation-induced changes in chemical shifts upon addition of ammonium 1 to solutions of 2. On the other hand, the new sets of signals seen in the 1H-NMR spectra correspond to conformer B. The dramatic up-field shift observed for the pyrrolic protons in B must be the result of the close proximity of the fullerene sphere suggesting that supramolecule [(1)·(2)] adopts a conformation in which the C60 subunit is located atop the porphyrin macrocycle. Further evidence for C60–porphyrin interactions came from UV–vis measurements. Addition of increasing amount of 1 to a CH2Cl2 solution of 2 causes a red shift of the Soret band, no further spectral changes being observed beyond the addition of ca. 5 equiv. of 1 (λmax = 421 and 427 nm before and after addition of 1, respectively). The latter observation confirmed the existence of the C60–porphyrin interactions in [(1)·(2)]. Effectively, red-shifts in the Soret band have been observed for covalent C60–porphyrin conjugates due to intramolecular π-stacking of the two chromophores [6]. Finally, the formation of supramolecular complex [(1)·(2)] was also evidenced in the gas phase by electrospray mass spectrometry (ES-MS). The spectrum obtained from an equimolar mixture of 1 and 2 displayed only one peak at m/z = 2587.2 corresponding to the 1:1 complex after loss of the trifluoroacetate counteranion (calculated m/z = 2587.24). The Ka value for the binding of 1 to 2 was determined by a fluorescence titration. A surprisingly high value for the association constant Ka = 3.75 × 105 M−1 was obtained. Effectively, the association constant between porphyrin 2 and fullerene 1 is unexpectedly increased by two orders of magnitude when compared to the Ka value found for the complexation of 1 with commercially available benzo-18-crown-6 (Ka = 2.1 × 103 M−1, determined in CDCl3 by NMR titration). Such a stabilization of the supramolecular complex formed between 1 and 2, which can be attributed to an additional intramolecular interaction, provides further evidence for the π-stacking of the fullerene moiety and the porphyrin subunit in [(1).(2)] suggested by the NMR studies. As depicted in Fig. 4, the calculated structure of supramolecular complex [(1).(2)] reveals that the linker between the ammonium group and the fullerene sphere has the perfect length to allow intramolecular stacking of the fullerene moiety and the porphyrin subunit in full agreement with the experimental observations.
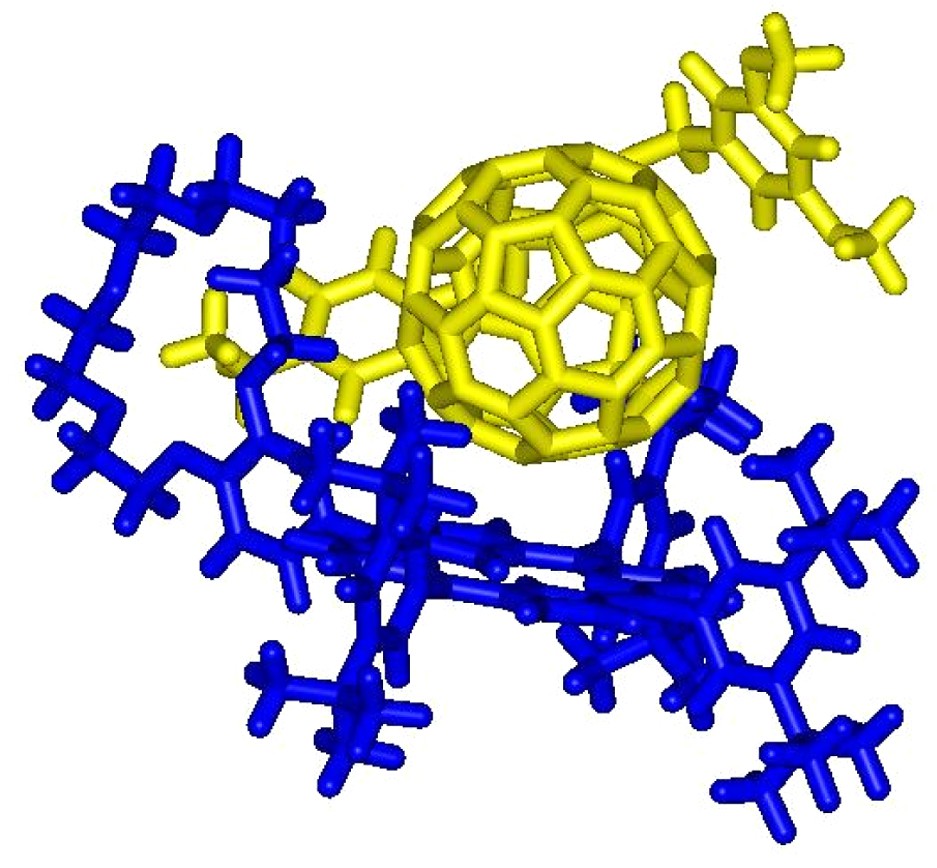
Calculated structure of the supramolecular complex [(1).(2)] (molecular modeling performed with Spartan, the dodecyl chains have been replaced by methyl groups in the calculations).
We have also shown that more complex supramolecular systems are easily accessible by using similar interactions [9] as illustrated by the association of cation 1 with porphyrin derivatives 3 and 4 bearing two crown ether subunits (Fig. 5).
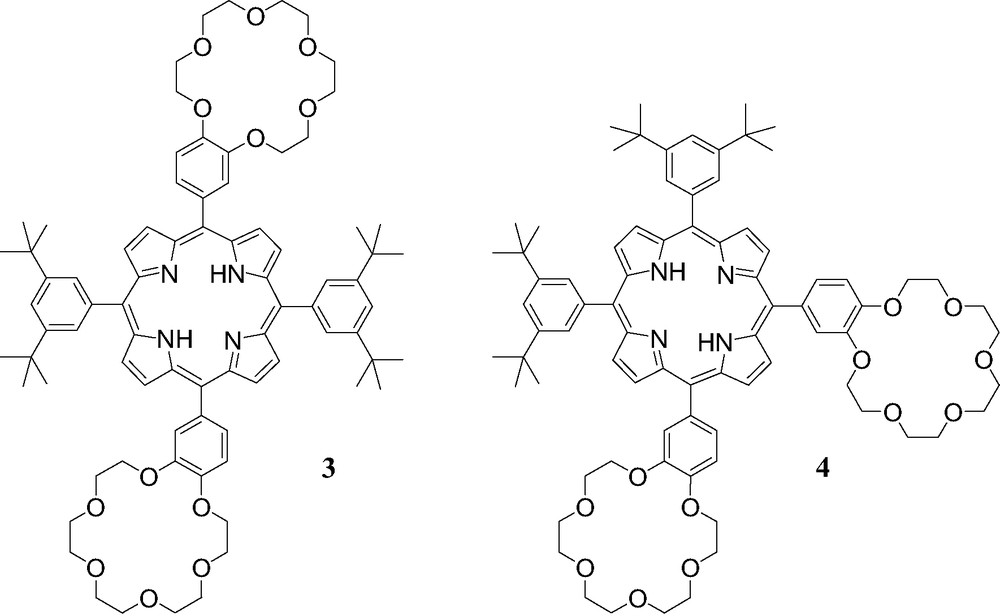
Compounds 3 and 4.
The binding behavior of 1 to porphyrins 3 and 4 was also investigated by 1H-NMR in CDCl3 at 298 K. As discussed for the NMR binding studies of 1 to 2, apparition of new sets of signals and complexation-induced changes in chemical shifts were observed in the 1H-NMR spectra of 3 and 4 upon addition of ammonium 1 revealing the effective formation of supramolecular structures. However, the interpretation of the NMR spectra appeared difficult due to the possible formation of 1:1 and 2:1 complexes. It must however be noted that, as in the case of [(1)·(2)], intramolecular C60–porphyrin interactions were evidenced for the supramolecular complexes obtained from the association of both 3 and 4 with ammonium 1. Effectively, some signals corresponding to the pyrrolic protons were dramatically up-field shifted in the 1H-NMR spectra of 3 and 4 upon addition of methanofullerene 1. Furthermore, by adding increasing amount of 1 to CH2Cl2 solutions of 3 and 4, the characteristic red shift of the Soret band due to the C60–porphyrin interactions was observed in the UV–vis spectra. The supramolecular complexes obtained from fullerene derivative 1 and the porphyrin–crown ether conjugates 3 and 4 were further characterized in the gas phase by ES-MS. As shown in Fig. 6, the positive ES mass spectrum obtained under mild conditions (extracting cone voltage Vc = 60 V) from a 2:1 mixture of 1 and 3 is characterized by a doubly charged ion peak at m/z = 2055.3 which can be assigned to the 2:1 complex after loss of the two trifluoroacetate counteranions (calculated m/z = 2055.45). The peak corresponding to the 1:1 complex could almost not be detected under these conditions, thus suggesting that the self-assembled array [(1)2.(3)] is the most abundant species in the analyzed solution.
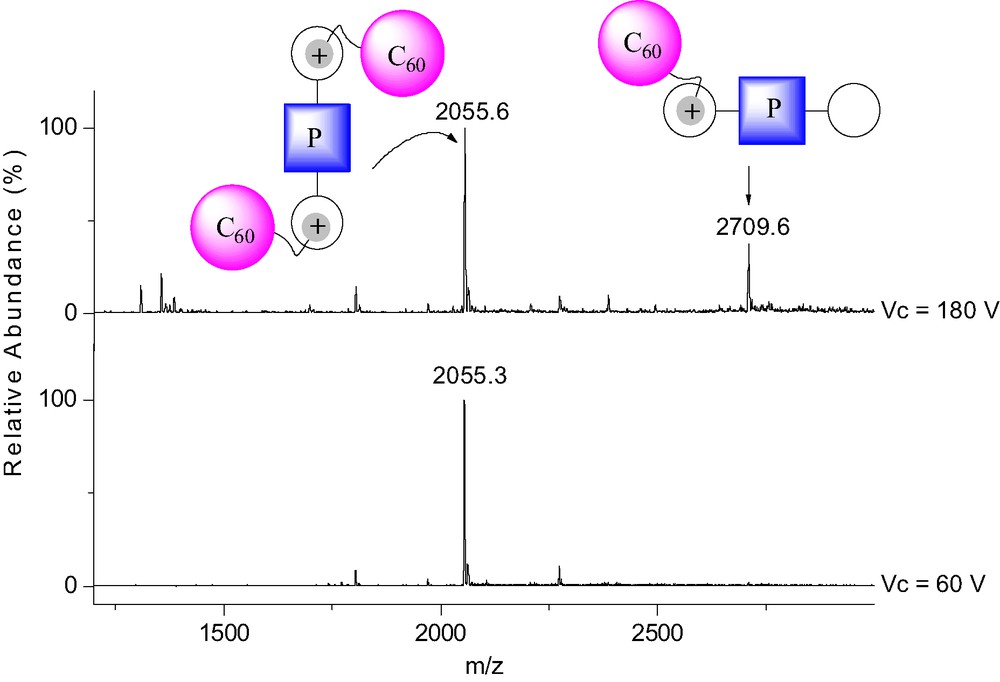
ES mass spectra recorded at different Vc from a 2:1 mixture of 1 and 3 in CH2Cl2.
Under more brutal conditions (Vc = 180 V), the spectrum was still dominated by the doubly charged ion peak at m/z = 2055.6 but a singly charged ion peak attributed to the 1:1 complex was also observed at m/z = 2709.6 (calculated m/z = 2709.28). The intensity of the latter signal is increased as the Vc value is increased, thus the peak corresponding to [(1)·(3)] must result from the fragmentation of the 2:1 supramolecular complex [(1)2·(3)] initially present in solution. A similar behavior was also observed for the ES mass spectra of the 2:1 mixture of 1 and 4 recorded at various Vc values. In both cases, the fragmentation is still limited due to the high stability of the non-covalent arrays [(1)2·(3)] and [(1)2·(4)]. This observation further corroborates the existence of intramolecular stacking between the fullerene moiety and the porphyrin subunit in [(1)2·(3)] and [(1)2·(4)] as suggested by the NMR and UV–vis data. Molecular modeling studies have been carried out. The calculated structures of [(1)2·(3)] and [(1)2·(4)] revealing intramolecular porphyrin–fullerene interactions are in full agreement with the experimental observations (Fig. 7).
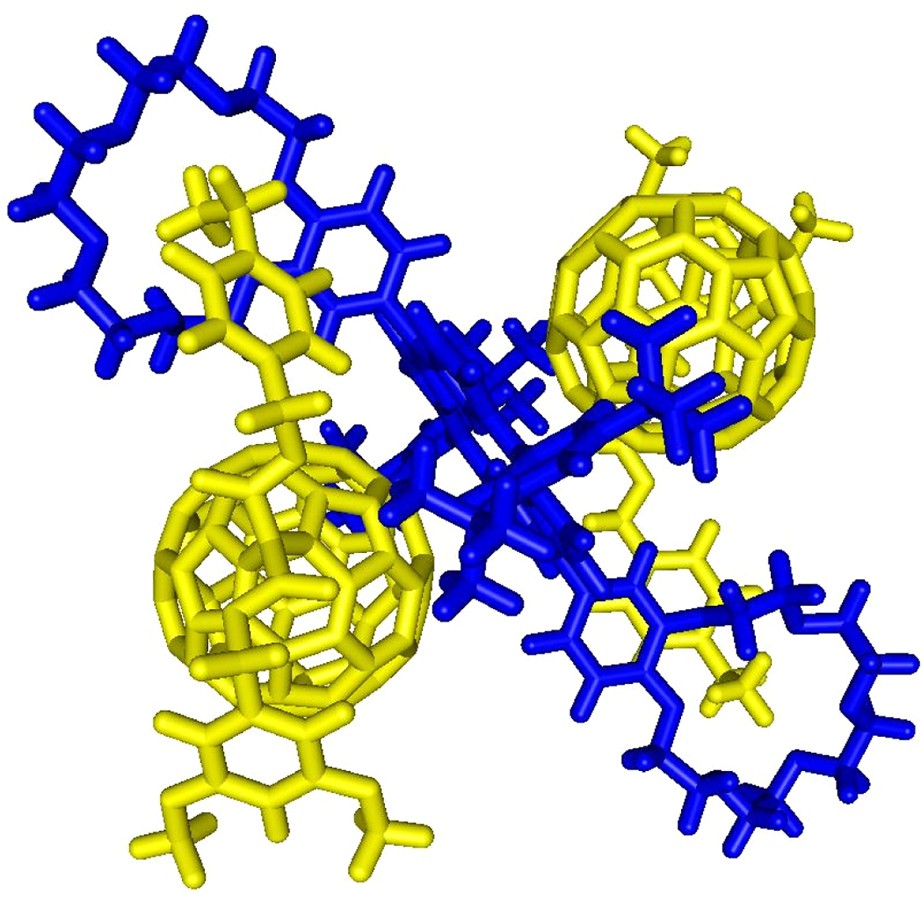
Calculated structure of the supramolecular complex [(1)2.(3)] (molecular modeling performed with Spartan, the dodecyl chains have been replaced by methyl groups in the calculations).
The complexation between 1 and 3 or 4 was finally investigated in CH2Cl2 by luminescence studies. A large decrease in intensity of the characteristic porphyrin emission was observed when the fullerene ammonium salt 1 was added to a CH2Cl2 solution of 3 or 4. The observed decrease in luminescence intensity originates from either energy or electron transfer from the photoexcited porphyrin to the C60 acceptor in the supramolecular complexes. Steady-state measurements are however not sufficient to determine the nature of the quenching, because the residual porphyrin emission overlaps the much weaker fullerene emission and so prohibits clean excitation spectroscopy. Detailed photophysical studies are currently under investigation and will be reported in due time.
3 Supramolecular oligophenylenevinylene–fullerene conjugates
Photochemical molecular devices based on the combination of C60 with π-conjugated oligomers have generated significant research activities in the past few years [10]. In particular, such donor-fullerene arrays have shown interesting excited state properties [10] and have been used as photovoltaic materials in solar cells [11]. This prompted us to study the complexation of methanofullerene 1 with the oligophenylenevinylene (OPV)–crown ether conjugate 5 (Fig. 8) [12]. The comparison between the 1H-NMR spectra of 1, 5 and a 1:1 mixture of both compounds revealed complexation-induced changes in chemical shifts for some protons, in particular for those of the 4-(aminomethyl)benzyl moiety in 1 and of the benzocrown ether unit in 5 (Fig. 8). The latter observations are in good agreement with the proposed structure of the supramolecular complex obtained from 1 and 5. The Ka value for the 1:1 complex deduced from the complexation-induced changes in chemical shifts of the 4-(aminomethyl)benzyl protons in 1H-NMR binding titrations (CDCl3, 298 K) was determined to be 3.5 × 103 M−1. The formation of the 1:1 complex [(1)·(5)] was also evidenced in the gas phase by ES-MS. The spectrum recorded from a 1:1 mixture of 1 and 5 displayed only one singly charged ion peak at m/z = 2471.4, which can be assigned to the 1:1 complex after loss of the trifluoroacetate counteranion (calculated m/z = 2471.3).
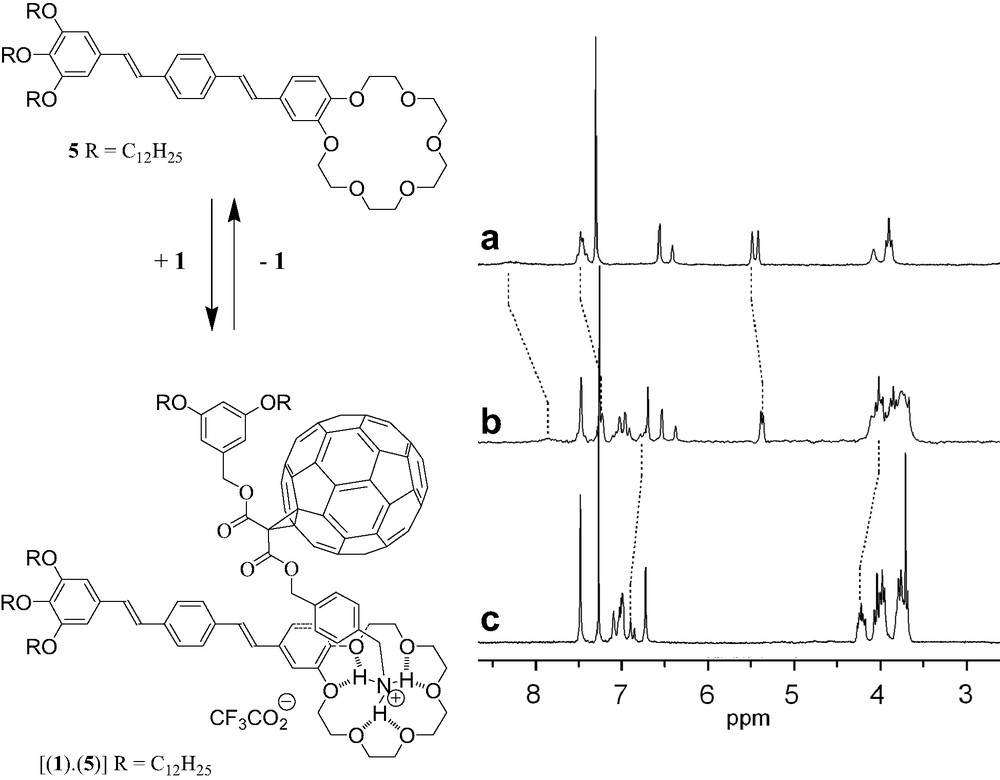
1H-NMR spectra (200 MHz, CDCl3) of 1 (A), 5 (C) and a 1:1 mixture of both compounds (B).
The complexation between 1 and 5 was also investigated in CH2Cl2 by luminescence studies. Evidence for an intramolecular quenching of the OPV excited state by the fullerene moiety in [(1).(5)] was obtained from the experiment depicted in Fig. 9. Addition of 1,4-diazabicyclo[2.2.2]octane (DABCO) (1.5 equiv.) to the 1:1 mixture of 1 and 5 in CH2Cl2 causes an increase of the OPV emission. Actually, the fluorescence intensity of the resulting solution was found to be similar to that of a reference solution containing 5 and methanofullerene derivative unable to form a complex with 5. In other words, the treatment with a base deprotonates the ammonium moiety of 1 and, thereby, disrupts the non-covalent bonding interactions that brought the components together. Addition of trifluoroacetic acid (2 equiv.) to the CH2Cl2 solution regenerates the ammonium center, thus allowing the formation of the supramolecular complex, and the luminescence intensity of final solution is the same as that of the starting 1:1 mixture of 1 and 5. The observed decrease in luminescence intensity originates most probably from energy transfer from the photoexcited OPV to the C60 acceptor in the supramolecular complex [(1)·(5)]. Finally, the association constant for the binding of 1 to 5 was also determined by a fluorescence titration in CH2Cl2. At this point, it must be emphasized that both intramolecular and intermolecular quenching processes occur upon addition of 1 to solutions of 5 in CH2Cl2. All the luminescence experiments were therefore carried out in the presence and in the absence of base in order to always have a suitable reference [12]. The corresponding Ka value was similar to the one deduced from the NMR binding studies.
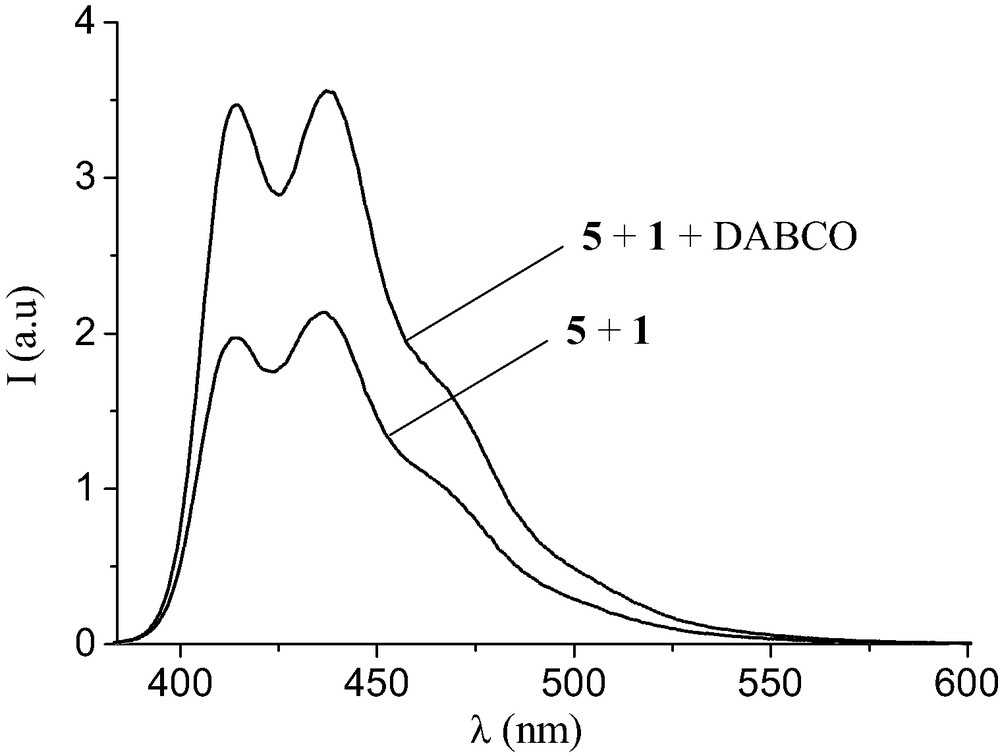
Emission spectra (λexc = 372 nm corresponding to the maximum of absorption of 5) of an equimolar mixture of 1 and 5 in CH2Cl2 before and after addition of DABCO (1.5 equiv.).
The non-covalent OPV–fullerene system [(1)·(5)] is a photoactive supramolecular device. However, the ammonium–crown ether interaction is the only recognition element responsible for the association of the two components and, in contrast to the complexation of 1 with the porphyrin guests 2–4, no additional π-stacking stabilization has been evidenced in this case. As a result, the binding constant found for the complex obtained from 1 and 5 is rather low and only a small fraction of the two components are effectively associated in solution. This encouraged us to design a new fullerene-ammonium derivative capable of providing additional recognition elements in order to increase the association constants. The Cs symmetrical fullerene bis-adduct 6 (Fig. 10) was prepared [13]. When compared to compound 1, additional phenyl units have been introduced to favor the occurrence of intramolecular π–π interactions in the supramolecular complex resulting from the binding of ammonium 6 to crown ether 5.
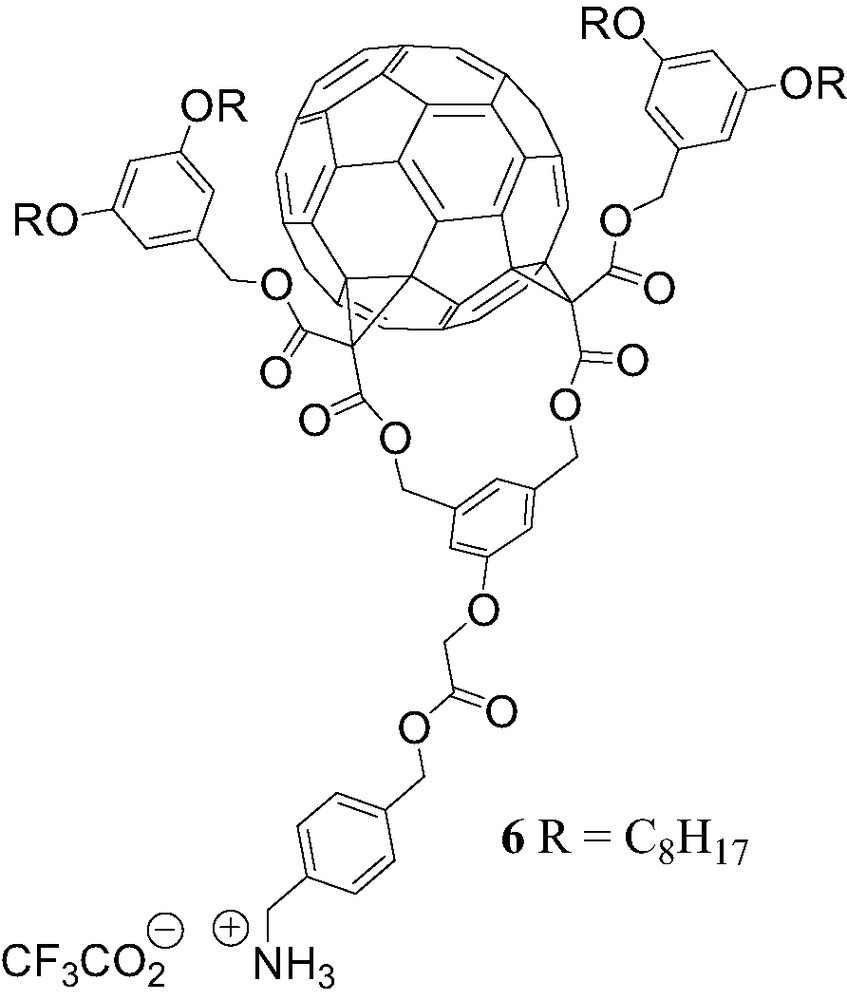
Compound 6.
The ability of fullerene derivative 6 to form supramolecular complexes with the crown ether 5 was first evidenced by ES-MS. The mass spectrum recorded from a 1:1 mixture of 5 and 6 displayed only one singly charged ion peak at m/z = 2983.8 assigned to the 1:1 complex after loss of the trifluoroacetate counteranion (calculated m/z = 2983.8). To quantify the interactions between the OPV-based host and the C60-ammonium guest, the complexation between 6 and 5 was further investigated in CH2Cl2 by fluorescence binding studies. The Ka value thus determined (Ka = 6.3 × 104 M−1) revealed a strong stabilization of the supramolecular complex. Effectively, the association constant between OPV 5 and fullerene 6 is increased by more than one order of magnitude when compared to the Ka value found for the complexation of 5 with cation 1. The later observation can be explained by a sum of secondary intramolecular weak interactions like π–π stacking or hydrophobic associations in [(5)·(6)].
The assembly of the C60-ammonium cation 6 with OPV derivative 7 bearing two crown ether moieties (Fig. 11) was also investigated [13]. ES-MS studies revealed the formation of the supramolecular complex [(6)2·(7)]. Effectively, the peak corresponding to this complex after loss of the counteranions was observed at m/z = 2417.8 (calculated m/z: 2417.9) in the mass spectrum obtained from a mixture of 7 (1 equiv.) and 6 (2 equiv.).
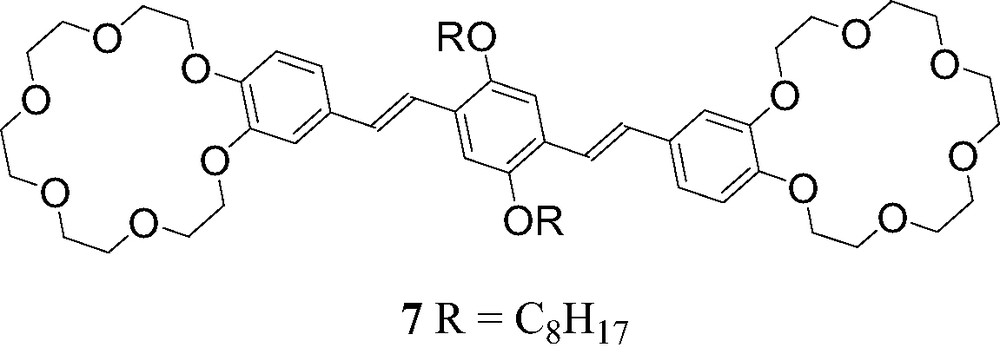
Compound 7.
Here again, we took advantage of the efficient quenching of the OPV emission by the C60 moiety upon complexation of 7 with 6 to determine the Ka values by luminescence titrations. In this case, the processing of the titration data led to the determination of two binding constants defined by equilibria (1) and (2):(1)
Values of 104 and 3.98 × 104 M−1 were found for K1 and K2, respectively. Interestingly, the ratio K2/K1 ≈ 4 is significantly larger than 0.25 which is the value expected for a statistical model of two identical binding sites [14]. The later observation clearly indicates that the stability of the supramolecular complex [(6)2·(7)] is significantly higher than that of its analogue [(6)·(7)] due to positive interactions. Our thermodynamic data suggest a structure in which the two C60 units of [(6)2·(7)] strongly interact through π–π stacking interactions as observed in the calculated structure of the 2:1 supramolecular complex depicted in Fig. 12.
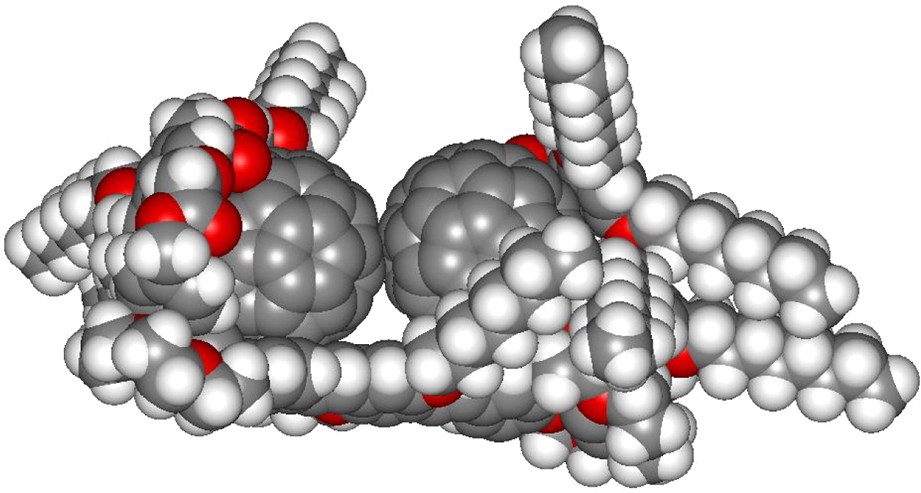
Calculated structure of the supramolecular complex [(6)2·(7)] (molecular modeling performed with Spartan).
Finally, the bis-ammonium fullerene ligand 8 (Fig. 13) was designed to form a 1:1 macrocyclic supramolecular complex with the bis-crown ether receptor 7 [15]. Molecular modeling studies revealed effectively a perfect complementarity of the two components (Fig. 13).
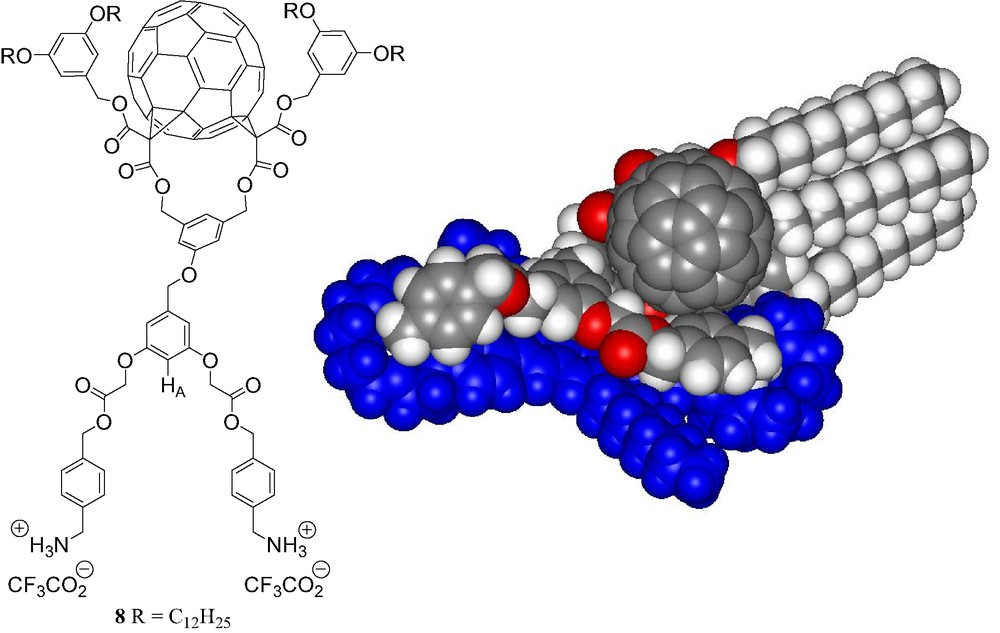
Compound 8 and calculated structure of the supramolecular complex [(7)·(8)] (molecular modeling performed with Spartan).
The binding behavior of bis-ammonium 8 to the bis-crown ether receptor 7 was first investigated by 1H-NMR in CDCl3 at 298 K. The comparison between the 1H-NMR spectra of 7, 8 and an equimolar mixture of both components revealed complexation-induced changes in chemical shifts. In particular, a dramatic down-field shift is seen for the signal of HA (see Fig. 13) in 8 upon addition of 7. The latter observation is in good agreement with the formation of a macrocyclic 1:1 supramolecular complex in which proton HA is located on top of the π-conjugated system of 7 (Fig. 13). The formation of a 1:1 complex was further evidenced by ES-MS. The positive ES mass spectrum (Fig. 14) recorded under mild conditions from a 1:1 mixture of 7 and 8 is dominated by a doubly charged ion peak at m/z = 1723.1 which can be assigned to the 1:1 complex without the trifluoroacetate counteranions (calculated m/z = 1723.11). A very minor peak corresponding to the 2:1 complex was also detected at m/z = 2226.7 (calculated m/z = 2226.76). By increasing the Vc value, the spectrum was still dominated by the doubly charged ion peak at m/z = 1723.1 and no significant fragmentation of the supramolecular assembly [(7).(8)] could be observed. Indeed, the spectra are almost identical regardless the Vc value. This behavior is in sharp contrast with the ES-MS analysis of related supramolecular complexes. Effectively, important fragmentation occurs at high Vc values and shows the high stability of the non-covalent array [(7)·(8)]. It is also worth noting that the signal of the supramolecule [(7)·(8)] remained the major peak in the ES mass spectra obtained from a 10:1 mixture of 7 and 8. Under these conditions, the intensity of the peak corresponding to the 2:1 complex [(7)2.(8)] which is lower than 5% when compared to the peak of the macrocyclic 1:1 associate, provides an additional evidence for the high stability of [(7)·(8)].
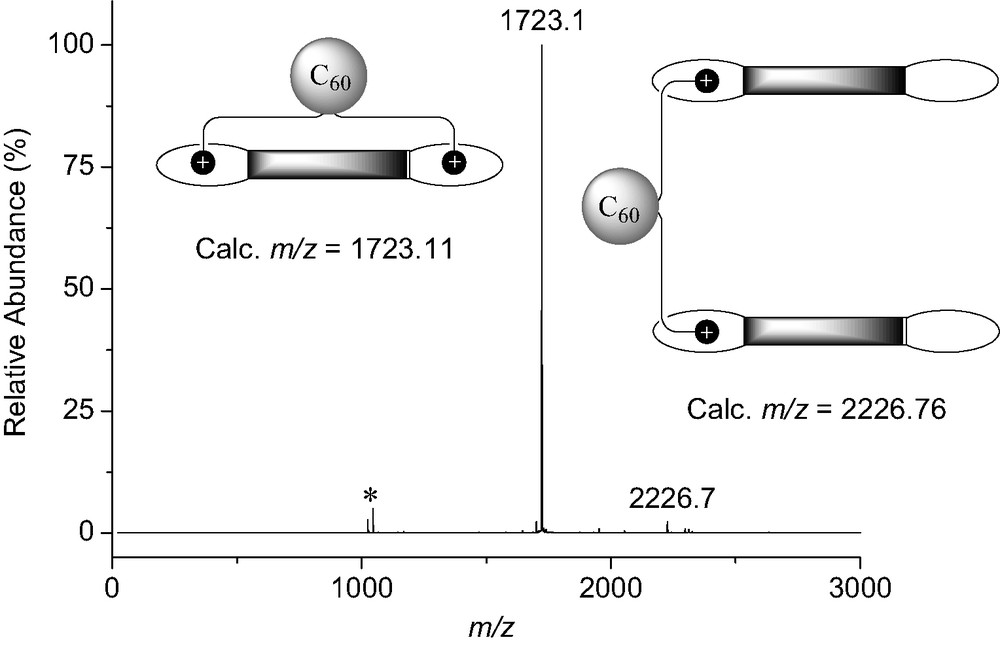
ES mass spectrum (Vc = 200 V) recorded from a 1:1 mixture of 7 and 8 in CH2Cl2 ([(7)] = [(8)] = 5 × 10−6 M); the peak at m/z = 1046.2 (*) is assigned to [7 + K]+ (calculated m/z = 1046.40).
As already discussed for related systems, the strong emission of the π-conjugated system of 7 is quenched upon binding of 8 by an efficient intramolecular process in complex [(7)·(8)], thus allowing to easily determine the binding constant by luminescence titrations (Fig. 15). The stability constant of [(7).(8)] was found to be 2.51 × 106 M−1. The comparison with the thermodynamic data of 1 or 6 to OPV 5 shows a coordination stronger by two to three orders of magnitude. This is mainly associated to the two-center host–guest topography.
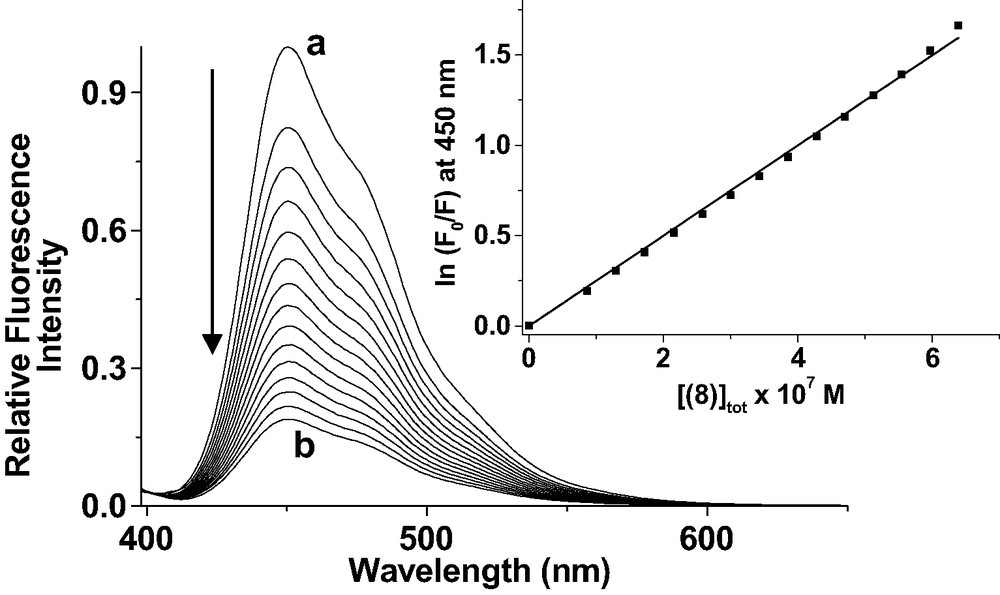
Luminescence titration of 7 by 8. Solvent: CH2Cl2; T = 25.0 ± 0.2 °C; λexc = 398 nm; emission and excitation slit widths 15 nm; 1% attenuator; (a) [(7)]tot = 9.77 × 10−7 M; (b) [(8)]tot/[(7)]to = 0.7. Inset: variation of ln(F0/F) at 450 nm versus the concentration of 8. The trend line is the result of the linear least-square fit of the experimental data according to ln(F0/F) = Ka[(8)].
4 Conclusion
In conclusion, we have shown that C60-ammonium derivatives and crown ethers functionalized with various chromophores are perfect building blocks for the preparation of fullerene-containing photoactive supramolecular systems. The ammonium–crown ether interaction itself is weak and the Ka values are rather low when the association of the two components results only from the binding of the cationic unit with the macrocyclic component. However, when additional recognition elements are present, the stability of the complexes is dramatically increased. In the case of the supramolecular porphyrin–fullerene conjugates, the additional intramolecular π-stacking between the two chromophores is responsible of the high stability of the self-assembled systems. Such attractive interactions have not been observed between the OPV moiety and the fullerene unit in the complexes obtained from the OPV–crown ether derivatives and the cationic fullerene guests. However, the phenyl substituents of the C60 derivative gives rise to intramolecular π–π interactions with the conjugated OPV moiety in the associate and thus, the resulting supramolecular complexes are stable. We have also shown that the assembly of a C60-ammonium cation with an OPV derivative bearing two crown ether moieties leads to the cooperative self-assembly of a stable 2:1 complex thanks to intramolecular fullerene–fullerene interactions. Finally, another strategy based on a two-center host–guest topography has been developed. Owing to the perfect complementarity of the two components, a bis-cationic substrate has been clicked on a ditopic crown ether derivative thus leading to a very stable non-covalent macrocyclic 1:1 complex. This approach is modular and appears easily applicable to a wide range of functional groups for the preparation of new supramolecular architectures with tunable structural and electronic properties.
Acknowledgements
This work was supported by the CNRS, the French Ministry of Research (ACI Jeunes chercheurs to N.S.), a doctoral fellowship from the French Ministry of Research to A.T., a doctoral fellowship from the CNRS and the Région Alsace to F.C., a doctoral fellowship from the GRK532 to H.H. and a post-doctoral fellowship from the Deutscher Akademischer Austausch Dienst (DAAD) to U.H.