

1 Introduction
Oxides and ozonides were among the earliest reported fullerene derivatives. Most of these compounds are quite easily obtained and purified. C70O was serendipitously discovered in 1991 in the toluene extract of arc-produced, fullerene-bearing ‘carbon soot’ [1]. Shortly afterward, Creegan et al. [2] reported the synthesis of C60O by photo-oxidation of C60 in oxygenated benzene, and Chibante and Heymann [3]1 reported the preparation of several C60 oxides through solution phase ozonation. These early investigations marked the start of fullerene oxide research.
Work in this area has been summarized in a 1997 paper by Taylor [4], and in a later review by Heymann [5] that includes studies through January of 2002. In this report, we attempt to survey relevant papers published through December 2005. We note that a number of fullerene compounds have been synthesized in which the carbon cage is derivatized with other groups in addition to oxygen. We choose to include here only such derivatives whose synthesis involves fullerene oxides or ozonides as starting materials. For example, C60F18O, which was obtained as a byproduct of the fluorination of C60 with K2PtF6 [6], is an oxygen-bearing fullerene that falls outside the scope of the present review.
The systematic chemical names of most fullerene derivatives are lengthy. For simplicity, we will refer here to compounds with formulas CmOn as Cm-oxide (for n = 1), Cm-dioxide (for n = 2) etc., and to Cm(O3)n compounds as Cm-ozonides, Cm-diozonides etc. When the –O– moiety of an oxide bridges a [6,6] C-C bond in the fullerene cage, the derivative will be called an epoxide to denote retention of the underlying C-C bond. Alternatively, when the oxygen atom bridges a [4,5] C–C bond, the underlying bond is weakened or broken to give an ether-like structure called an annulene. In the case of higher oxides (n > 1), combinations of epoxide and annulene sites are possible. The process of reacting with ozone will be termed ozonation, and fullerene derivatives containing O3 addends will be called ozonides.
Section 2 describes methods for preparing and purifying fullerene oxides and ozonides. Section 3 deals with their qualitative and quantitative analysis. Section 4 treats the stability of oxides and ozonides in solution both in darkness and when exposed to light. Section 5 discusses theoretical studies and molecular structures. Finally, other observations are briefly treated in Section 6.
2 Sources and purification of oxides and ozonides
Fullerenes do not react readily with molecular oxygen in the absence of energetic electromagnetic radiation [7–12]. However, their oxides form as byproducts of fullerene synthesis in the carbon-arc process or in oxygen-starved combustion of hydrocarbons. These processes yield soot from which oxides have been solvent-extracted [1,13–18].
Photo-oxidation of C60 in O2-saturated benzene was the process used in the first direct synthesis of C60O [2,19–22]. This reaction is slow and yields only the [6,6]-epoxide. Schuster et al. [23] suggested that this photochemical synthesis proceeds via reaction of singlet molecular oxygen with the lowest triplet excited state of C60. A failed attempt was made to synthesize the [5,6]-open oxidoannulene isomer by the photoxidation of C60 in oxygenated benzene [24].
Ozonation of fullerenes is a frequently used method for the synthesis of ozonides and oxides [25–49]. In this method, ozone gas is bubbled through solutions of fullerenes. Reaction is rapid, and the ozone dosage must be carefully monitored because exposure to excess ozone leads to abundant insoluble substances and destruction of recognizable fullerenes [3,50–54]. Polyketones and esters are also among the main stable products [55]. Although fullerene ozonides, the initial reaction products, are not stable, their lifetimes are generally long enough to permit purification by high performance liquid chromatography (HPLC). Fullerene ozonides decompose by losing molecular O2 to form fullerene oxides that differ in structure depending on whether the ozonide dissociation was thermolytic or photolytic [38–40,45,46,49]. Gas phase ozonations have also been carried out in mass spectrometers to produce and study large odd-numbered carbon clusters [56,57].
Alternative oxidants that have been used to produce fullerene oxides in solution reactions are dioxiranes [58–60], m-chloroperoxybenzoic acid [20,61–71], cytochrome P450 [72] and methyltrioxorhenium/hydrogen peroxide [73]. Oxidation under ultrasonication was carried out with 3-chloroperoxy benzoic acid, 4-methyl morpholine N-oxide, chromium(VI) oxide, oxone(R)monopersulfate, 3-picoline N-oxide, pyridine N-oxide hydrate, quinoline N-oxide, and isoquinoline N-oxide [74–76]. For a variety of objectives, oxides or their ions were also produced in gas phase studies [7,10,25,26,29,43,56,57,61,77–83]. Syntheses using reactants in the solid state have been mainly performed to prepare fullerene multimer oxides such as C120O [35,61,84–86], but these compounds have also been produced in solution [32,45,46,87,88]. A rather unusual synthesis of C120Om compounds was carried out by the decomposition of C60Br24 in air [89]. Separations and purifications of mixtures of oxides and ozonides are nearly always done using HPLC. Details of those methods can found in many of the above-cited papers. However, an elegant kinetics-based method for the effective pre-separation of fullerene oxides is available when the lifetimes of two or more ozonide precursors differ greatly [39,40].
3 Qualitative and quantitative analyses
The main methods used for qualitative and quantitative analyses are HPLC, mass spectrometry (MS) and nuclear magnetic resonance spectrometry (NMR). In principle, qualitative analysis of crystallized substituted oxides can also be performed through X-ray diffraction (XRD) studies, although this is a laborious approach [62–68]. Infrared spectra have been reported for several oxides [2,12,17,19,20,41,58,85,89–91], but such spectra have seldom been used for identification.
HPLC analysis can detect the formation of new, previously unknown substances formed through chemical reactions, but it cannot establish their chemical formulae or structures. To obtain such information, data from MS, NMR, or XRD are generally necessary. Once the formula and electronic absorption spectrum of an ozonide or oxide are known, HPLC serves as the simplest technique for monitoring its presence. Table 1 lists references to reported UV–vis spectra of fullerene oxides and ozonides. HPLC has the advantage of being a non-destructive technique because samples are usually recoverable. However, its utility is compromised when regioisomers are insufficiently resolved, or when an analyte has an unknown absorption spectrum or a very long retention time.
Published UV–vis spectra
| Compound | References |
| C60 adducts | |
| [6,6]C60O epoxide | [2,3,15,49,53,63,92] |
| [5,6]C60O annulene | [49] |
| C60O2 dioxide | [34,46,65] |
| C60O2 dioxide I | [40] |
| C60O2 dioxide II | [40] |
| C60O2 dioxide III | [40] |
| C60O3 trioxide | [46,58] |
| C120O | [14,77,86] |
| C120O2 | [45,46,85] |
| C120On | [87] |
| C60(O3) ozonide | [38] |
| C60(O3)2 diozonide I | [40] |
| C60(O3)2 diozonide II | [40] |
| C60(O3)2 diozonide III | [40] |
| C60O(O3) oxyozonide I | [40] |
| C60O(O3) oxyozonide II | [40] |
| C60O(O3) oxyozonide III | [40] |
| C70 adductsa | |
| C70O | [1,13,20,67,93]b |
| C70O 5 regioisomers | [39] |
| C140O | [87] |
| C70(O3) a,b and c,c ozonides | [39] |
a Equivalent carbon atoms of the C70 cage are labeled a through e from the pole to the equator.
b Computationally predicted spectrum.
For quantitative HPLC analyses, the molar absorptivity of a given compound must be known for at least one wavelength in the detection range. Beck et al. [26] reported determining absolute molar absorptivities for C60O but did not report the numerical values. Although Fusco et al. [59] had sufficient masses of C60O, C60O2, and C60O3 to determine their solution phase absorptivities, the values seem not to have been reported. Richter et al. [16] used ‘approximate’ but unspecified absorptivities for C60O and C70O. To our knowledge, the only numerical values published in the open literature are for the C60O epoxide (41,000 dm3 mol–1 cm–1 at 328 nm) [49,92], the C60O annulene (42,000 dm mol–1 cm–1 at 336 nm) [49], and C120O2 (115,000 dm3 mol–1 cm–1 at 329 nm) [45,46]. Because the yield of C60O epoxide from thermolysis of the C60O3 ozonide is nearly 100%, it is possible to deduce the absorptivity of the ozonide as 44,000 dm3 mol–1 cm–1 at 325.8 nm [38]. Note that all of these wavelengths and absorptivities are somewhat solvent-dependent.
Various mass spectrometric methods have been used for qualitative and very approximate quantitative analysis of oxides. Unfortunately, ozonides appear too unstable for mass spectrometric studies, and MS cannot distinguish between isomers such as the epoxide and annulenes forms of C60O. MS techniques that have been used in published reports include laser desorption time-of-flight (both with and without matrix), electron impact (with samples introduced in volatile solvents), atmospheric pressure chemical ionization, fast atom bombardment, electrospray ionization, and thermospray [1,2,13,17,18,30–33,35,47,48,69,70,72,84,85,88].
13C NMR spectra of fullerene oxides may be measured for natural abundance samples, although limited solubility in NMR-compatible solvents often leads to long data acquisition times. For fullerene ozonides, NMR spectroscopy faces the additional challenge of sample instability. Consequently, an NMR spectrum is available only for the c,c-C70(O3) ozonide [39]. Table 2 lists three aspects of the reported NMR spectra. The complete set of line positions in the sp2 region is available in most of the cited papers. Structural implications of these NMR spectra will be considered in Section 5.
Published NMR spectra
| Compound | Number of sp2 signals | sp2 chemical shifts (ppm) | sp3 chemical shifts (ppm) | References |
| C60 compounds | ||||
| C60O-epoxide | 15 | 140–146 | 90.18 | [2] |
| C60O-epoxide | 15 | 141.99–146.32 | 90.92 | [58] |
| C60O-epoxide | 15 | 140.89–145.33 | 90.02 | [72] |
| C60O-annulene | 38 | 133.39–151.99 | [49] | |
| C60O2-diepoxide | 139–150 | 78.4; 82.3 | [94] | |
| C60O2-diepoxide | 26 | 139.19–149.57 | 78.46; 82.40 | [72] |
| C60O2-diepoxide | 30 | 139.6–149.8 | 78.5; 82.5 | [69] |
| C60O2-diepoxide | 30 | 139.65–149.84 | 78.61; 82.55 | [59] |
| C60O3-trioxide† | 49 | 138.19–150.37 | 65.88; 69.06; 73.08; 75.29; 76.96; 80.19; 82.61 | [59] |
| C120O | 32 | 136.2–153.7 | 78.9; 90.0 | [88] |
| C120O2b | 27 | 137.58–149.28 | 72.19; 92.11 | [84] |
| C120O2(A), major | 95 | 135.993–154.612 | 77.797; 78.839; 80.197; 85.361; 93.552; 99.880 | [35] |
| C120O2(A), minor | 13 | 135.737–155.347 | 72.410; 77.596; 78.991; 80.420; 92.352; 98.370; 98.867; 104.302 | [35] |
| C120O2 | 56 | 135.496–151.669 | 82.251; 91.455 | [46] |
| C70 compounds | ||||
| c,c-C70(O3) ozonidec | 29 | 140.65–153.86 | 97.02 | [39] |
| C70O mix† | 72 | 130–153 | 85.20; 90.41; 91.84 | [1] |
| a,b-C70O epoxidea | 35 | 130.85–153.61 | 90.99; 92.42 | [13] |
| a,b-C70O epoxide | 35 | 130.60–153.37 | 90.73; 92.17 | [39] |
| a,b-C70O epoxidea | 35 | 130.92–151.61 | 85.78 | [13] |
| a,a-C70O annulenec | 40 | 135.33–155.49 | (No data reported) | [39] |
| C140O(A) | 35 | 131.26–162.32 | 70.07; 94.36 | [87] |
| C140O(B) | 127 | 125.38–162.90 | 67.99; 72.95; 90.59; 94.79 | [87] |
a Mix of two isomers;
b Synthesized by the dimerization of [5,6]-C60O annulenes;
c The paper reported only those lines that were not obscured by solvent features; some lines may have arisen from reaction products formed during measurement.
4 Stability
4.1 Thermolysis
This section deals with thermolysis and photolysis of the known fullerene oxides and ozonides. Little has been reported to date on the thermal stability of the oxides in solution2 [95]. Fullerene ozonides, however, decompose detectably in the dark at room temperature [38–40]. Table 3 lists activation energies (Ea), exponential decay lifetimes at a specific temperature, and the principal products of ozonide decompositions. All reactions follow first order kinetics. The thermal dissociation of these fullerene primary ozonides (1,2,3-trioxolanes) is slow compared to that of other organic primary ozonides. This probably reflects the steric impossibility of rearrangement to give 1,2,4-trioxolane structures, which are commonly formed from primary ozonides in other systems but in fullerene adducts would require penetration of oxygen atoms into the fullerene cage. Note that the rate constant for C60(O3) decomposition is much higher in toluene than in octane solution at the same temperature. For C60(O3), a,b-C70(O3), and for a mixture of the I, II, and III isomers of C60(O3)2, it is known that thermolysis produces O2 [38–40]. Reaction channels with the unresolved simultaneous loss of two O2 molecules were not found among the diozonides. Furthermore, the C60O and a,b-C70O products are epoxides but the d,d-C70O product is an annulene. C60O2 dioxide I and C60O2 dioxide II in Table 3 are identical, with two previously reported diepoxides [15,34,66]; C60O2 dioxide III is, most likely, also a diepoxide [40].
Decomposition of ozonides and oxido-ozonides in toluene and octane
| Ozonide | Ea (kJ mol–1) | Exponential lifetime (min) | Products | References |
| C60(O3) | 89 | 22 at 296 K | C60O; O2 | [38] |
| C60(O3)a | 794 at 296 K | C60O | [38] | |
| C60(O3)2 I | 52 ± 5 at 288 K | C60O(O3) I; O2b | [40] | |
| C60O(O3) I | 69.7 ± 0.7 at 289.6 K | C60O2 I | [40] | |
| C60(O3)2 II | 62 ± 6 at 288 K | C60O(O3) II; O2b | [40] | |
| C60O(O3) II | 58 ± 6 at 289.6 K | C60O(O3) III; O2b | [40] | |
| C60O(O3) III | 87 ± 21 at 288 K | C60O2 III | [40] | |
| a,b-C70(O3) | 94.3 | 14 at 296 K | a,b-C70O; O2 | [39] |
| c,c-C70(O3) | 102 | 650 at 296 K | d,d-C70Oc | [39] |
a In octane; all other entries are for decomposition in toluene;
b O2 release was observed from the combined ozonides;
c [5,6]-open annulene.
4.2 Photolysis and photoisomerization
A systematic investigation revealed that C60(O3) in toluene was photolyzed very readily to form O2 and the C60O annulene isomer [49], with essentially no formation of the C60O epoxide isomer that is the exclusive product of C60(O3) thermolysis. A failed attempt was reported to synthesize this C60 isomer by the direct photoxidation of C60 in oxygenated benzene [24]. When exposed to 324 nm radiation at 24 °C in air-saturated toluene, the photochemical quantum yield of C60(O3) was found to be approximately 6% [49]. Fig. 1 illustrates the remarkably distinct pathways for thermal and light-induced dissociation of C60 ozonide. Subsequent studies revealed that all known fullerene ozonides and some oxides undergo photochemical reactions. Those processes found for C70 are graphically presented in Fig. 2. Table 4 summarizes the photoproducts of C60(O3), C70(O3) isomers, and C70O annulenes. The four C70O annulenes were found to photoisomerize to form two epoxides that have much higher photostability [39]. These isomerizations involved migrations of the oxygen atom on the surface of the C70 when the fullerene was in its triplet electronic state. Studies of the photolysis of C60(O3)2 found three dioxide products of undetermined structures [40]. One important practical consequence of these facile photoinduced reactions is that fullerene ozonations carried out in light produce more product species than ozonations carried out in the dark.
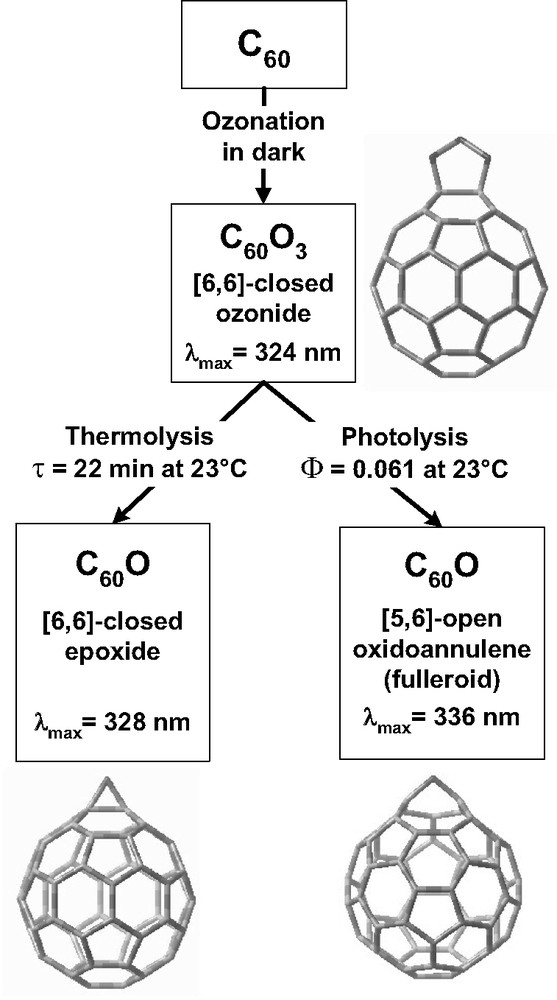
Reaction pathways and product characteristics for the formation of C60O isomers by the solution phase ozonation of C60.
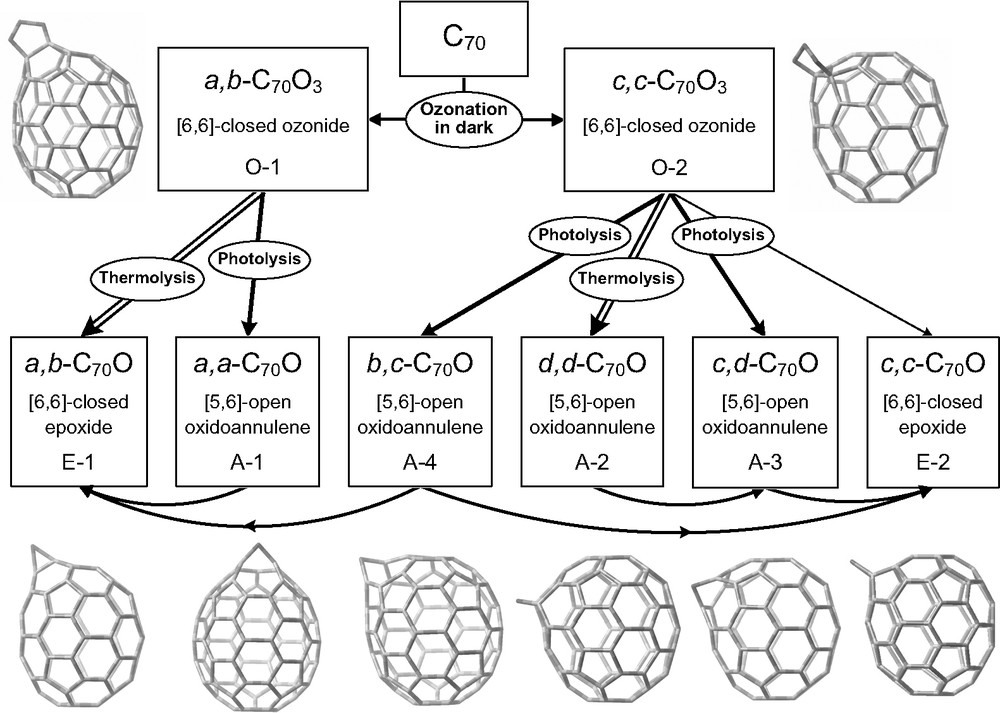
Photo and thermal reaction pathways and deduced product structures for C70O isomers formed by the solution phase ozonation of C70.
Products of photolysis of C60(O3) and C70(O3) ozonides and C70O oxides
| Ozonide or oxide | Photoproduct(s) | References |
| C60(O3) ozonide | [5,6]-open C60O annulene | [49] |
| a,b-C70(O3) ozonide | [5,6]-open a,a-C70O annulene | [39] |
| c,c-C70(O3) ozonide | [5,6]-open b,c-C70O annulene; [5,6]-open c,d-C70O annulene; [6,6]-closed c,c-C70O annulene | [39] |
| [5,6]-open a,a-C70O annulene | [6,6]-closed a,b-C70O annulene | [39] |
| [5,6]-open b,c-C70O annulene | [6,6]-closed a,b-C70O annulene; [6,6]-closed c,c-C70O annulene | [39] |
| [5,6]-open d,d-C70O annulene | [5,6]-open c,d-C70O annulene | [39] |
| [5,6]-open c,d-C70O annulene | [6,6]-closed c,c-C70O annulene | [39] |
5 Structure determination through experiment and theory
This section brings together facts and ideas about the structures of fullerene ozonides and oxides. For single-cage derivatives, one aims to understand whether the cage contains any open C–C bonds and how and where the addends are bonded to the cage. For multi-cage derivatives, one also seeks to understand the covalent linkage between cages.
5.1 Ozonides
Release of O2 indicates that the –O–O–O– moiety of all ozonides is bonded to the outside of the cage. However, although computations and UV-vis spectroscopy on C60O3 suggest that the O3 group straddles a [6,6]-closed C–C bond, no conclusive experimental evidence for such ozonide structures is yet available. The observation that thermolysis of C60(O3) yields the C60O epoxide cannot be accepted as proof because thermolysis of c,c-C70(O3) is known to produce an annulene. However, stability and kinetics calculations predict that [6,6]-closed ozonide adducts are more likely to form than [5,6]-open isomers [41,95,96]. There is currently no direct evidence concerning the regiostructures of the three C60(O3)2 diozonides. Because each decomposed to a single C60O(O3) oxyozonide intermediate, however, the O3 groups must have been attached to the cages on either side of a plane of symmetry. Heymann et al. [40] assigned probable structures to the three diozonides on the basis of calculated dipole moments of all eight possible regioisomers and observed HPLC retention times, which are known to depend on analyte polarity. The mechanism of reaction of C70 with ozone and possible structures of C70O3 ozonides were studied theoretically [97].
5.2 C60O epoxide and annulene
Creegan et al. [2] demonstrated using NMR data that the C60O they had synthesized by photooxygenation was the [6,6]-closed epoxide isomer. They pointed out that its UV–vis spectrum retained the essential features of the C60 spectrum. However, the UV–vis spectrum of the annulene is actually much more similar to that of C60: it shows a near-UV peak at 336 nm, lacks the sharp 424 nm band present in the epoxide, and has broad visible peaks at wavelengths close to the corresponding features of C60. This electronic similarity between the [5,6]-open isomer of C60O and pristine C60 reflects the retention of 60 electrons in the annulene's π-system, in contrast to the 58 π-electrons in C60O epoxide [49]. Theoretically calculated values were reported for energies and structures, including bond lengths and pyramidalization angles, of both isomers [35,60,96,98–103]. These results confirmed that the epoxide has a lower energy than the annulene. Additional structural confirmation of the [6,6]-closed adduct was obtained by the unique X-ray diffraction analysis of (η2-C60O)Ir(CO)Cl(As(C6H5)3)2 and (η2-C60O)Ir(CO)-Cl(P(C6H5)3)2 crystals, which ‘visualized’ the C60O epoxide structure [63,64,68].
5.3 C50O
The eight possible isomers of this compound have been studied only through quantum chemical computations. They indicate that the most stable C50O isomer is an annulene structure with oxygen bridging across the [5,6]-open bond between the pole and the equatorial belt [104].
5.4 C60O2 dioxides
Six C60O2 dioxides were prepared from diozonides, three by thermolytic and three by photolytic decomposition [40]. Here we distinguish these by the wavelengths (in nm) of their characteristic near-UV absorption maxima: C60O2(314), C60O2(316), C60O2(320), C60O2(328), C60O2(332), and C60O2(333). A compilation of all available theoretical and experimental structural information revealed that two regioisomers, C60O2(320) and C60O2(316) (isomers I and II of reference [34]), are diepoxides, that C60O2(314) is probably another diepoxide, and that the remaining isomers are either diannulenes or epoxyannulenes [26,34,42,59,69,72,94,103,105].
5.5 C60O3 trioxide
13C NMR spectra were used to identify two reported isomers of C60O3 trioxide: 1,2:3,4:9,10-triepoxy [60]fullerene and 1,2:3,4:11,12-triepoxy [60]fullerene [72].
5.6 C120O dimer
Although several investigators have studied C120O, only one NMR-based study has clearly shown that the C60 cages are connected by a furanoid (C4O) bridge [88]. The spectrum shows that this species has C2v symmetry. Energies and NMR spectra were theoretically calculated for four possible C120O isomers, and the results confirmed that the C2v-symmetry isomer lies lowest in energy [106]. Crystalline C120O-metal-octaethylporphyrin was characterized by X-ray diffraction, but there was sufficient disorder to prevent determination of the bridge structure [107].
5.7 C120O2 dimers
Four different isomers have been synthesized and characterized by MS, UV–vis, IR, and 13C NMR spectroscopies. One isomer has C2v symmetry with the C60 cages linked by two adjacent furanoid rings [35,84]. Two closely related isomers have a C60 moiety linked through a single furanoid ring to a C60O epoxide moiety [35]. This so-called A-isomer has 16 possible regioisomers. Calculations suggest that the epoxy O-atom is bonded in a cis-1, e, or trans position relative to the furanoid bridge [35].
The fourth isomer was synthesized via spontaneous room-temperature dimerization of the [5,6]-open annulene isomer of C60O. The most likely of four possible isomers for this species was found to be the nonpolar dimer with C2 symmetry, which has a basic cage structure of dumbbell-shaped C120 with each O-atom bridging a [5,6]-open C–C cage bond adjacent to the sp3 carbons that link the two C60 cages [45,46]. Remarkably, it was observed that this C120O2 isomer undergoes photodissociation to regenerate the C60O annulene from which it was formed. This unique chemical system can therefore provide mixtures of a fullerene monomer and its dimer in a dynamic balance that can be readily controlled by adjusting concentration, temperature, and optical irradiation [46].
5.8 C180O2 trimer
By heating a mixture of C60, C60O, C60O2, and C60O3, Deng et al. [32] synthesized a compound that had a mass corresponding to the formula C180O2. They suggested that its structure consisted of three C60 cages held together by two furanoid rings in a V-shaped arrangement.
5.9 C70O monoxides
Diederich et al. [1], who discovered C70O, suggested a [6,6]-open annulene structure that turned out to be incorrect. Several theoretical calculations indicated that the most stable form of C70 monoxide was the e,e (‘equatorial-belt’) isomer [96,100,108], while others indicated the a,b and c,c isomers were more stable [109,110]. Experimental results either from the XRD structures [62,67] or NMR spectroscopy demonstrated that the a,b and c,c isomers are preferentially formed, although it was unclear whether thermodynamic or kinetic factors were dominant [13,20,39]. Heymann et al. [39] synthesized and separated six C70O regioisomers through C70 ozonation followed by thermolysis or photolysis. Two of the resulting C70O products were identified as epoxides and four as annulenes on the basis of their UV–vis spectra, HPLC retention patterns, and an NMR spectrum of the a,a annulene.
5.10 C140O and C140O2
Three distinct C140O isomers were isolated and then investigated with NMR [87]. Although the structural assignments could not be totally firm, the investigators preferred the C(2)–O–C(2), C(2)–O–C(3), and C(1)–O–C(2) structures in which the C70 cages are held together by furanoid rings and the numbers in parentheses index the carbon atom site on each cage to which the oxygen atom is bonded. The symmetries of these preferred structures are C2v, C1, and Cs, respectively. Prior to the experimental study, calculated NMR signatures and energies for seven C140O and four C140O2 isomers were reported [106]. It was predicted that three of the seven possible C140O isomers have nearly equal energies. In addition, the energies of all four of the C140O2 isomers were calculated to be very similar.
6 Other observations
Several chemical reactions have been reported in which fullerene ozonides and oxides participate as reactants. Ozonides decompose to oxyozonides and oxides plus molecular oxygen [38–40,49]. Fullerenes and fullerene oxides have been reacted with one another to synthesize dimeric oxides [35,45,46,84,85,87,88,111,112]. The facile synthesis of Vaska-type Ir complexes with C60O, C70O, and C84O has greatly contributed to structural studies of fullerene oxides [62–65,67,68].
The use of MS, and especially laser desorption methods, for determining the mass of fullerene oxides led to the discovery of novel fragmentation and coalescence processes [25,26,61,77]. The odd-numbered carbon clusters C53, C55, C57, C59, C119, C129, and C139 were discovered, usually as negative ions, as products of gas phase reactions involving molecular oxygen or ozone and fullerene oxides, or simply through decomposition of oxides [7,29,43,56,57,79]. It was suggested that C119 has a spirane-like structure [113], and the proposed structure was consistent with theoretical calculations [114].
Photophysical studies on C60O epoxide revealed an anomalous temperature dependence of the triplet state decay lifetime [92]. This phenomenon may be related to the facile triplet state photoisomerization that interconverts isomers of C70O by allowing the oxygen addend to ‘walk’ on the fullerene surface [39]. The electrochemical behavior of C60O in solutions shows that the first and third electrons are added to the fullerene cage, whereas the second electron is added at the epoxide oxygen, resulting in a dianion that initiates polymerization [71].
The stable fullerene oxides and their derivatives can be obtained in the solid state by simple evaporation of solvents. C60O forms an orientationally disordered face-centered cubic lattice at room temperature. Upon cooling, an orientational ordering transition occurs to a simple cubic phase [21,115]. A ‘glass transition’ similar to the one observed in C60 has also been reported for C60O [44]. Stable poly(fullerene oxide) films have been prepared by thermal activation and electron bombardment of hexa-nitro [59] fullerene films on a gold substrate. The films were reportedly stable up to 900 K [116,117].
Acknowledgements
The authors are grateful for research support from the National Science Foundation (grant CHE-0314270) and the Welch Foundation (grant C-0807). We also thank our co-workers and collaborators for their important research contributions.
1 The first ozonation of C60 in toluene was performed in 1992 by J. W. Verhoeven and D. Heymann in the Laboratory for Organic Chemistry of the University of Amsterdam. Dr. Verhoeven declined co-authorship on Ref. [3].
2 Heymann et al. reported (Ref. [40]) that the three synthesized C60 ozonide diepoxides remained unchanged in room temperature toluene for at least three weeks.