

1 Introduction
Calcium phosphates have been widely studied for dentistry and orthopedic implants because of their high biocompatibility and bioresorbability [1–3]. Hydroxyapatite derivatives (Ca5(PO4)3(OH)) (including carbonated apatitic phases) give bones, teeth and tendons their hardness and stability. By “playing” with the chemical composition, selective protein adsorption properties can be obtained for nanocrystalline hydroxyapatite [4]. Apatitic materials may also act as drug delivery systems [5–8].
Solid-state NMR is definitely suitable for the in-depth characterization of calcium phosphate phases. 31P NMR has been largely used in the past, in combination with 1H → 31P Cross-Polarization transfer [9]. Moreover, inorganic components in bones were investigated [10]. 1H solid-state NMR has been rarely used [11] due to the lack of resolution related to the strong homogeneous 1H–1H dipolar interaction. Multiple pulses experiments combined with MAS (CRAMPS) [12] have been implemented for the characterization of calcium phosphate phases [13]. Nevertheless, full assignment of proton lines remains difficult, though empirical trends correlating the isotropic chemical shifts and the H-bond networks have been proposed in the literature some decades ago [14]. In this paper, we demonstrate that first-principles calculations are validated for 31P solid-state NMR. Calculated CSA tensors are in very good agreement with experimental data obtained by MAS or static experiments. When considering 1H MAS spectra (at very high field and very fast rotation frequency), calculated data become essential for full assignment of the experimental spectra. All calculations were done by using the GIPAW method first developed by Mauri and Pickard (see Section 2) [15].
17O solid-state NMR experiments appear as a straightforward tool of characterization of the local structure of inorganic materials [16] by probing directly and quantifying the different types of oxo-bridges present in the structure. The 17O isotropic chemical shift values of M–O–M′ fragments (M, M′ = Si, P, Ti, Zr,…) are indeed highly sensitive to the chemical nature of M and M′ [17–19]. However, the main drawback of this technique is its poor sensitivity, due to the low natural abundance of the 17O isotope (0.037%). In the field of phosphate compounds, this problem can nonetheless be overcome either by using 17O-enriched water for the hydrolysis step of the precursors [20] or by heating the final material under 17O-enriched water vapour [21] or 17O2 gas [22]. Despite isotopic enrichment and high-resolution MQ-MAS (Multiple-Quantum Magic-Angle Spinning [23]) methods, the interpretation of 17O spectra still remains difficult, as no empirical rules have yet been found to enable the assignment of the peaks and to obtain structural information for each site. In this context, first-principles calculations can be particularly useful in the understanding of spectra, since accurate calculation of the NMR parameters will allow the assignment of all sites.
43Ca (I = 7/2, 0.145%) NMR remains exotic in inorganic chemistry due to very low sensitivity and large quadrupole moment. It must be noticed that recent work dealing with model compounds and 43Ca NMR characterization at high field has been carried out in the literature by using large amount of samples [24].
The outline of the article is the following: Section 2 presents the syntheses of the studied calcium phosphate phases, including protonated structures (CaHPO4·2H2O, Brushite and Ca(H2PO4)2·H2O, MCPM) and non-protonated structures (β-Ca(PO3)2 and Ca4P2O9, TTCP). The first-principles calculations and the GIPAW approach are also described in Section 2. Section 3 deals with results and discussion: the calculated and experimental 31P are first discussed for all structures, including polymorphs of Ca(PO3)2. 1H NMR data are discussed for protonated phases, emphasizing the calculation of CSA tensors and the strong influence of H bonding. Finally, 17O predicted data (isotropic chemical shifts and quadrupolar constants) are summarized and discussed.
2 Experimental
Brushite, CaHPO4·2H2O (Fig. 1), was synthesized by mixing calcium carbonate and H3PO4 (85%) in water (Ca/P = 1). The mixture was stirred at room temperature until precipitation. For Ca/P = 0.5, MCPM, Ca(H2PO4)2·H2O (Fig. 2), was obtained. By heating MCPM at 600 °C (10 h), β-Ca(PO3)2 (Fig. 3) was synthesized. TTCP, Ca4P2O9 (Fig. 4), was obtained by mixing calcium carbonate and monetite (CaHPO4) at 1400 °C for 10 h, followed by quenching in air. The obtained compounds were systematically checked by powder X-ray diffraction (Brushite, JCPDS: 72-1240; MCPM, JCPDS: 70-0090; β-Ca(PO3)2, JCPDS: 11-0039; TTCP, JCPDS: 70-1379).
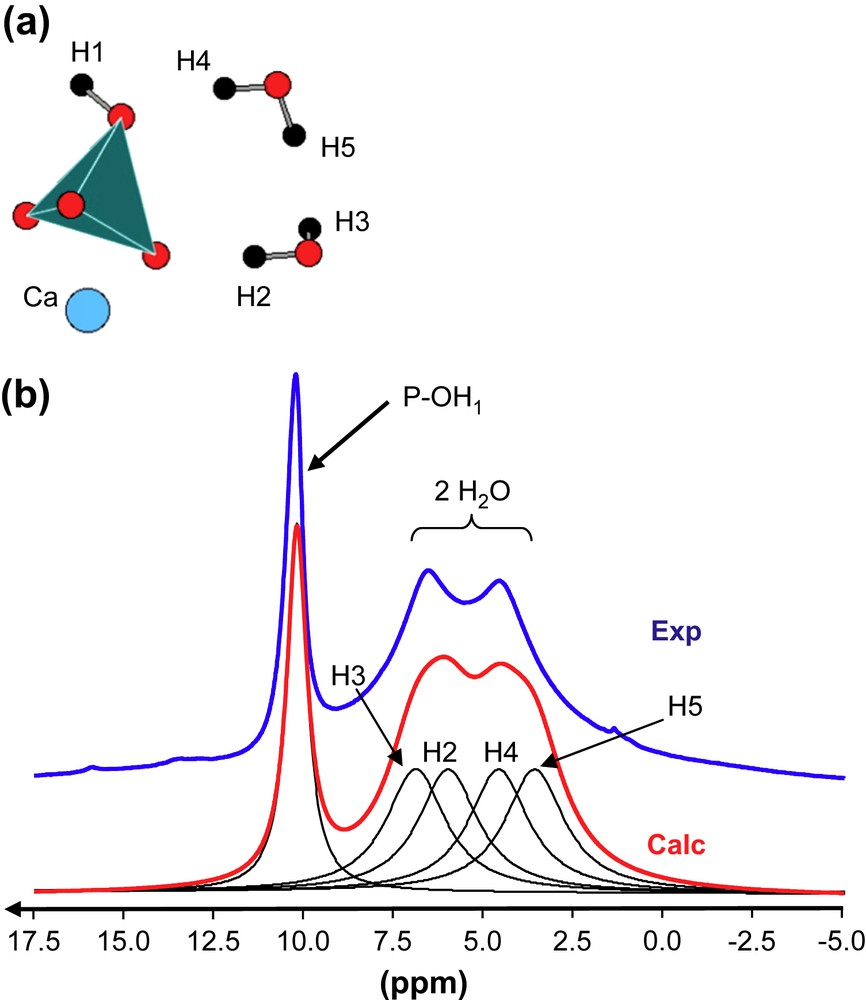
(a) Schematic representation of atoms involved in brushite. (b) 1H MAS NMR spectra (experimental and calculated) of brushite (17.6 T, 34 kHz). The calculated spectrum is obtained by using the GIPAW values given in Table 1.
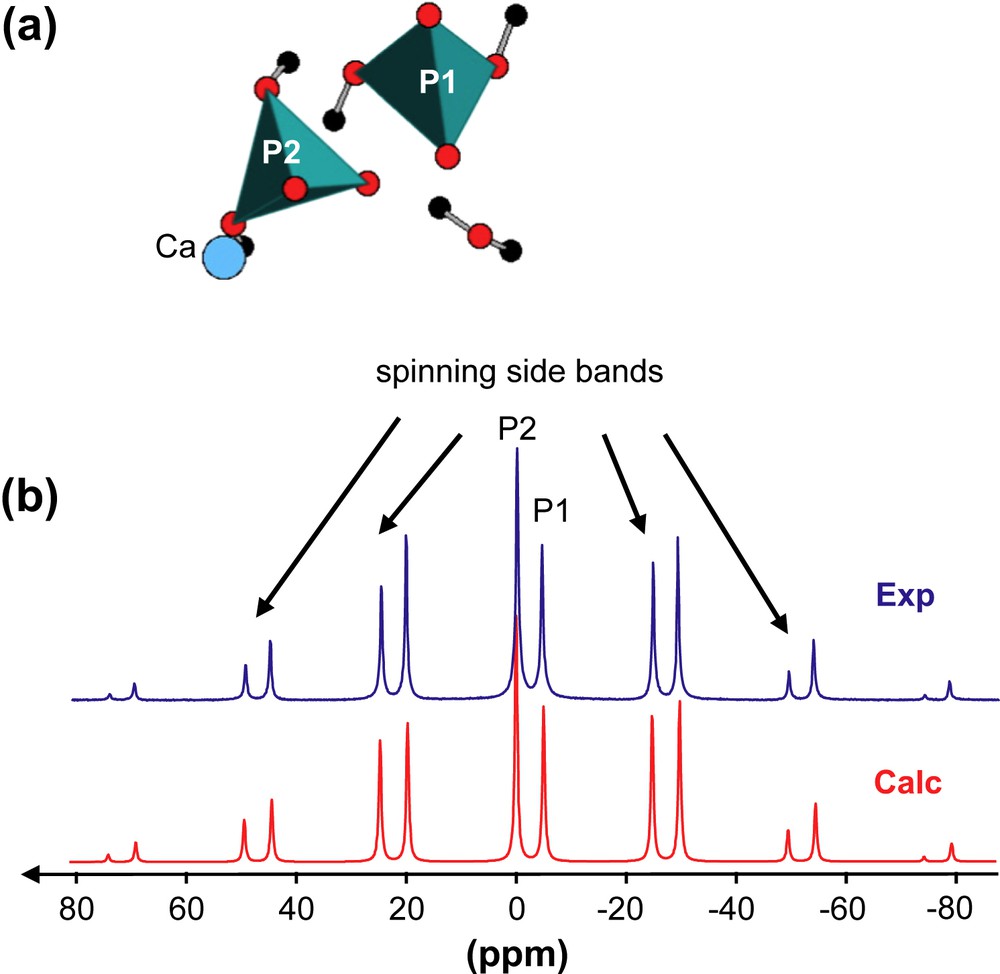
(a) Schematic representation of atoms involved in MCPM. (b) 31P MAS NMR spectra (experimental and calculated) of MCPM (7 T, 3 kHz).
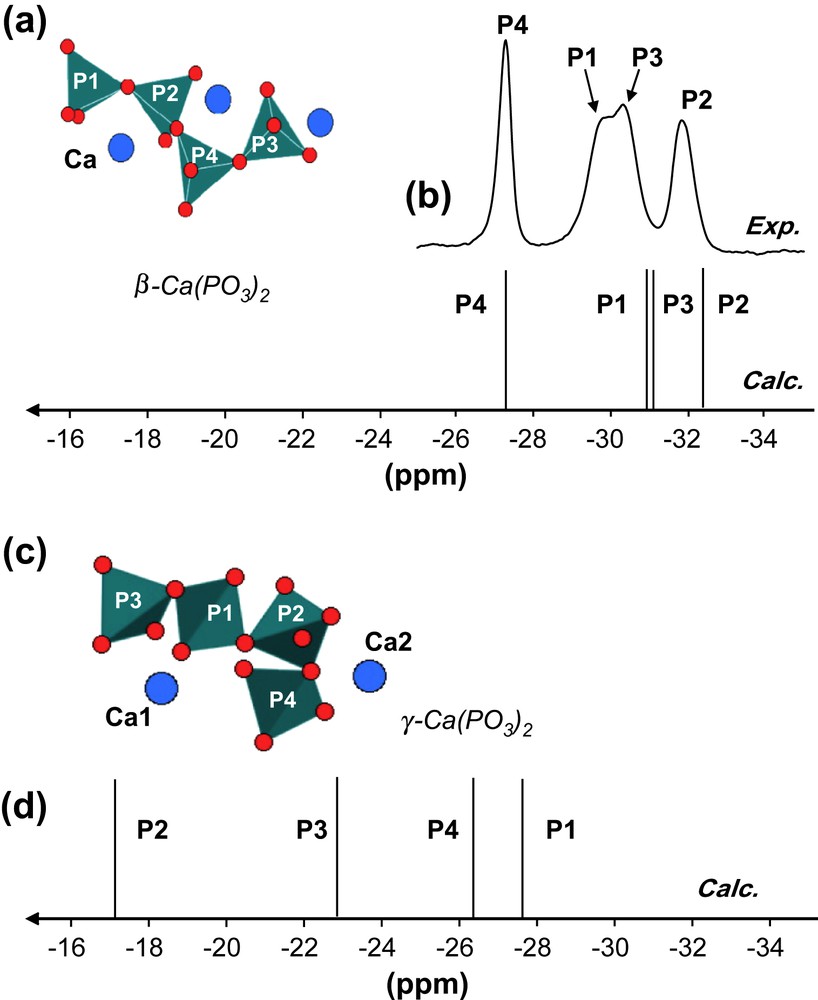
(a) Schematic representation of atoms involved in β-Ca(PO3)2. (b) 31P MAS NMR spectra (experimental and calculated) of β-Ca(PO3)2 (7 T, 14 kHz). (c) Schematic representation of atoms involved in γ-Ca(PO3)2. (d) 31P MAS NMR spectrum (calculated) of γ-Ca(PO3)2 (7 T, 14 kHz).
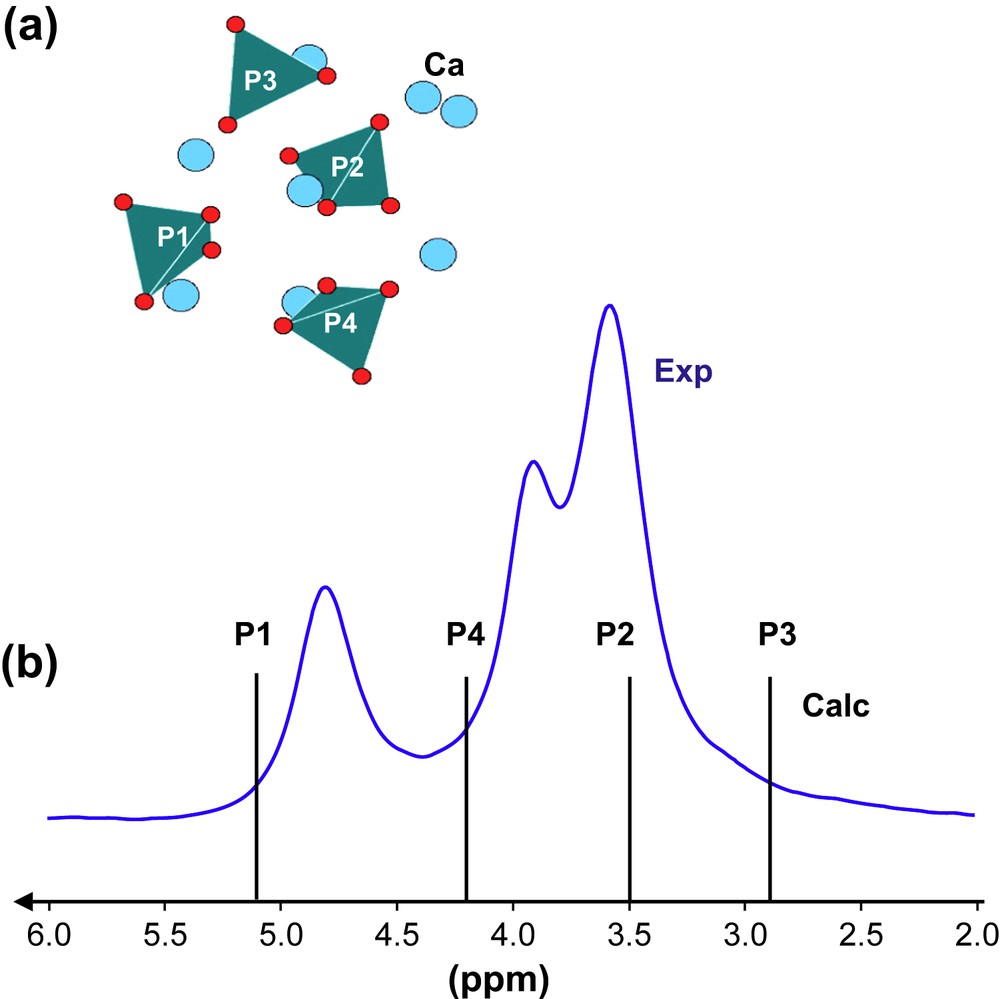
(a) Schematic representation of atoms involved in TTCP. (b) 31P MAS NMR spectra (experimental and calculated) of TTCP (7 T, 14 kHz).
1H NMR experiments were recorded on a Bruker AVANCE 750 spectrometer in order to enhance resolution (1H: 750.16 MHz). A zirconia rotor (2.5 mm) was spun at 33 kHz at the magic angle. The number of scans (NS) was 4, the pulse length was 1.7 μs (30°), and the relaxation delay (RD) was 30 s. The spectra were referenced to TMS. 31P MAS NMR experiments were performed on a Bruker AVANCE 300 (31P: 121.44 MHz) using a 4-mm Bruker probe under 1H high power decoupling (TPPM pulse sequence) (pulse length: 1.6 μs (30°), RD: 30 s). Spectra were referenced to H3PO4 (85%). A zirconia rotor (4 mm) was spun at 3 and 14 kHz.
The first-principles calculations were performed within the Kohn–Sham DFT. The PBE generalized gradient approximation [25] was used and the valence electrons were described by norm-conserving pseudopotentials [26] in the Kleinman–Bylander [27] form. The core definition for O is 1s2 and it is 1s22s22p6 for P and Ca. The core radii are 1.2 a.u. for H, 1.5 a.u. for O and 2.0 a.u. for P and Ca. The wave functions are expanded on a plane wave basis set with a kinetic energy cut-off of 80 Ry. The crystalline structure is described as an infinite periodic system using periodic boundary conditions. The NMR calculations were performed for the experimental geometries determined by neutron or X-ray diffraction for the various calcium phosphates: brushite, CaHPO4·2H2O [28]; MCPM, Ca(H2PO4)2·H2O [29]; β-Ca(PO3)2 [30]; γ-Ca(PO3)2 [31]; Ca4P2O9 [32]. In the case of Ca4P2O9, a significant better agreement is obtained between experimental and calculated 31P parameters by taking into account a geometry optimization (starting from the experimental geometry and allowing the positions of the atoms to relax using DFT calculations). The new coordinates obtained after relaxation are summarized in Supporting information. The integral over the first Brillouin zone was performed using a Monkhorst–Pack 2 × 2 × 2 k-point grid [33] for the charge density and electric-field gradient calculation and a 4 × 4 × 4 k-point grid for the chemical shift tensor calculation. The calculations have been performed at the IDRIS supercomputer centre of the CNRS using a parallel IBM Power4 (1.3 GHz) computer; the calculation of the Electric Field Gradient (EFG) and CSA tensors requires from 2 to 10 h on 32 processors. The shielding tensor is computed using the GIPAW approach which permits the reproduction of the results of a fully converged all-electron calculation, while the EFG tensors are computed using a PAW approach. The isotropic chemical shift δiso is defined as δiso = −[σ − σref] where σ is the isotropic shielding and σref is the isotropic shielding of the same nucleus in a reference system. In our calculations, absolute shielding tensors are obtained. The σref values were chosen by comparison between experimental and calculated δiso in a series of SiO2 polymorphs [34a] for 17O and other organophosphorus derivatives for 1H and 31P: σref (17O) = 261.5 ppm, σref (1H) = 31.0 ppm, and σref (31P) = 300.7 ppm. Diagonalisation of the symmetrical part of the calculated shielding tensor provides its principle components δ11, δ22 and δ33 defined such as |δ33 − δiso| ≥ |δ11 − δiso| ≥ |δ22 − δiso| and δiso = 1/3(δ11 + δ22 + δ33). Similarly, the principle components Vxx, Vyy, and Vzz of the EFG tensor defined as |Vzz| ≥ |Vxx| ≥ |Vyy| are obtained by diagonalisation. The quadrupolar interaction can then be characterized by the quadrupolar coupling constant CQ (with its sign) and the asymmetry parameter ηQ defined as: CQ = eQVzz/h and ηQ = (Vyy − Vxx)/Vzz. It has to be noticed that the role of Ca–O bonds within the frame of GIPAW calculations has been carefully studied by Profeta et al. [34b]. It was demonstrated that an easy and transferable correction of the PBE–DFT theory was possible for the accurate calculations of NMR parameters.
3 Results and discussion
3.1 31P: calculated vs. experimental data
Brushite, CaHPO4·2H2O, is characterized by a unique P crystallographic site (Fig. 1a). Static experiments (not shown here) allowed to extract the three principle components of the 31P CSA tensor (Table 1). Experimental and GIPAW calculated values are in very good agreement for all components. In the case of MCPM, two P sites are involved (Fig. 2a). The corresponding low-frequency 31P MAS NMR spectrum (at 3 kHz) is presented in Fig. 2b. The calculated spectrum is again in very good agreement, when considering both the 31P isotropic chemical shifts and the full CSA data (Table 1). The experimental CSA is characterized by the relative intensities of the spinning sidebands and can be extracted by using the DMFit program [35]. The two isotropic chemical shifts are separated by ∼5 ppm. This difference seems sufficient for safe assignment of the resonances: P1 corresponds therefore to the most shielded resonance.
Calculated 1H, 17O and 31P NMR parameters for brushite CaHPO4·2H2O, MCPM Ca(H2PO4)2·H2O, β-Ca(PO3)2, γ-Ca(PO3)2, and Ca4P2O9
| δiso (ppm) | CQ (MHz) | ηQ | δ11 (ppm) | δ22 (ppm) | δ33 (ppm) | ||
| Calc | Exp | Calc | Calc | Calc (exp) | Calc (exp) | Calc (exp) | |
| Brushite | |||||||
| P1 | 2.0 | 1.6 | −59.7 (−54.2) | −3.5 (−5.6) | 69.1 (64.7) | ||
| H1 (POH) | 10.2 | 19.9 | 18.1 | −7.4 | |||
| H2 (H2O) | 6.0 | 15.9 | 15.2 | −13.0 | |||
| H3 (H2O) | 6.9 | 17.0 | 15.7 | −12.0 | |||
| H4 (H2O) | 4.6 | 14.1 | 13.1 | −13.3 | |||
| H5 (H2O) | 3.5 | 11.6 | 9.3 | −10.3 | |||
| O (POH) | 75.5 | 6.9 | 0.9 | ||||
| O (PO⋯Ca) | 100.9–116.6 | 4.6–4.8 | 0.0–0.3 | ||||
| O (H2O) | −11.0 and −21.1 | 7.7 and 9.0 | 0.8 and 0.9 | ||||
| MCPM | |||||||
| P1 | −4.1 | −4.2 | −71.0 (−73.4) | −7.0 (−4.2) | 66.0 (65.0) | ||
| P2 | 0.8 | 0.3 | −50.0 (−51.2) | −4.0 (−2.3) | 55.5 (54.4) | ||
| H1 (POH) | 12.2 | 24.0 | 22.4 | −9.9 | |||
| H2 (POH) | 10.4 | 22.2 | 19.8 | −10.7 | |||
| H3 (POH) | 8.8 | 19.4 | 17.2 | −10.2 | |||
| H4 (POH) | 9.9 | 21.3 | 19.6 | −11.1 | |||
| H5 (H2O) | 6.4 | 16.9 | 14.8 | −12.6 | |||
| H6 (H2O) | 5.1 | 11.9 | 11.1 | −7.7 | |||
| O (PO⋯Ca) | 91.4–99.4 | 4.5–4.7 | 0–0.1 | ||||
| O (POH) | 64.5–74.9 | 6.7–7.5 | 0.7–0.8 | ||||
| O (H2O) | −0.9 | 7.6 | 0.8 | ||||
| β-Ca(PO3)2 | |||||||
| P1 | −30.8 | −29.9 | 67.8 (61.3) | 3.5 (−3.4) | −163.9 (−147.4) | ||
| P2 | −32.3 | −31.8 | 66.6 (58.9) | 10.1 (2.5) | −173.7 (−157.0) | ||
| P3 | −30.9 | −30.3 | 65.4 (60.8) | −3.2 (−3.9) | −155.0 (−148.0) | ||
| P4 | −27.3 | −27.3 | 57.2 (52.7) | −2.6 (−7.3) | −136.4 (−127.4) | ||
| O (PO⋯Ca) | 97.4–117.3 | 4.4–4.8 | 0.1–0.3 | ||||
| O (POP) | 100.1–118.2 | 8.1–8.3 | 0.2–0.4 | ||||
| γ-Ca(PO3)2 | |||||||
| P1 | −29.3 | 74.1 | 9.9 | −172.0 | |||
| P2 | −17.2 | 68.6 | 11.3 | −131.5 | |||
| P3 | −22.7 | 75.2 | 11.8 | −155.1 | |||
| P4 | −26.3 | 78.1 | 10.1 | −167.2 | |||
| O (PO⋯Ca) | 91.8–109.3 | 4.5–5.0 | 0.1–0.3 | ||||
| O (POP) | 102.7–130.3 | 7.6–7.9 | 0.4–0.5 | ||||
| Ca4P2O9 | |||||||
| P1 | 5.1 | 22.1 | 6.2 | −12.7 | |||
| P2 | 3.5 | 24.5 | 9.9 | −23.8 | |||
| P3 | 2.9 | 15.4 | 11.2 | −17.8 | |||
| P4 | 4.2 | −4.2 | 0.8 | 16.3 | |||
| O (PO⋯Ca) | 108.7–158.7 | 4.1–6.0 | 0.1–0.3 | ||||
| O (Ca⋯O) | 294.7–306.8 | 0.7–0.9 | 0.1–0.8 |
The structure of β-Ca(PO3)2 is presented in Fig. 3a. It contains chains of cornerlinked phosphate groups running in an unidirectional manner through the structure. Four P crystallographic sites are involved and four resonances are indeed observed in the corresponding 31P MAS NMR spectrum (Fig. 3b). The chemical shift difference between the less and most shielded resonances is ∼5 ppm. These resonances are consequently assigned to P2 and P4, respectively (Table 1). The assignment of the remaining resonances (P1 and P3) has been achieved by combining calculated data and 31P/31P MAS-J-INADEQUATE [36a] experiments [37]. Recently [31], a new polymorph of Ca(PO3)2 (namely γ-Ca(PO3)2) has been described in the literature. Chains of cornerlinked phosphate groups are also present, exhibiting different geometries when compared to the β-Ca(PO3)2 chains (Fig. 3c). The calculated 31P isotropic chemical shifts are obviously deshielded when compared to β-Ca(PO3)2 (Fig. 3d). Such results are in good agreement with 31P experimental data presented in Ref. [31].
The results obtained for brushite, MCPM, β- and γ-Ca(PO3)2 tend to validate the GIPAW calculations of 31P CSA parameters. The 31P MAS NMR spectrum of TTCP (Fig. 4b) shows overlapping isotropic resonances in a range less than 2 ppm. The four calculated lines (corresponding to the four P crystallographic sites – Fig. 4a) lie obviously in the same range. At this stage, it seems impossible to assign safely each line. As monophosphate groups are involved, experiments involving 31P/31P dipolar recoupling [36b] should help for full assignment of the MAS spectrum.
3.2 1H: calculated vs. experimental data
Ab initio calculations of 1H parameters are rarely described [38]. In most cases, 1H data are calculated within the cluster approximation [39], with some exceptions involving mainly organic molecules [40]. Only δiso (1H) values are discussed. It should be noticed that empirical correlations involving the δiso (1H) values and the internuclear distances have been previously described in the literature [14]. The goal of this section is to revisit such correlations from the first-principles calculation point of view.
Fig. 1b presents the 1H MAS NMR spectrum of brushite obtained at very high field (B0 = 17.6 T) and very fast rotation frequency (up to 34 kHz). Full resolution is not attained due to the strong 1H/1H dipolar interaction, which is homogeneous in nature [41]. However, three resonances located at ∼4.0, 6.5 and 10.0 ppm are observed. Calculated GIPAW values are summarized in Table 1, leading to the calculated spectrum shown in Fig. 1b. The most deshielded line corresponds obviously to POH groups. The other resonances are safely assigned to protons belonging to water molecules. In a sense, calculations allowed full assignment and interpretation of the MAS spectrum. Calculated 1H data related to MCPM are also given in Table 1.
Fig. 5 presents all calculated δiso (1H) values vs. the shortest OH⋯O distances (extracted mainly from neutron diffraction data). Distinct ranges for H2O and POH resonances are evidenced. Moreover, for a given range, deshielding of protons associated with a shortening of the OH⋯O distances is clearly demonstrated. Recently [42], such results have been extended to 1H data related to phenylphosphonic and phenylphosphinic acids. The 1H CSA data shown in Table 1 are consistent with nearly axial symmetry for all tensors (H2O and POH). Such data are in agreement with results already published in the literature [43]. Nevertheless, Wu et al. [44] have pointed out that 1H CSA tensors could deviate from axial symmetry in the case of crystalline hydrates (involving perchlorate and sulphate derivatives). Fig. 6 presents the absolute orientations of 1H CSA tensors in the case of MCPM. For POH and H2O protons, the unique axis (δ33) of the CSA tensors is nearly collinear to the OH⋯O bond direction.
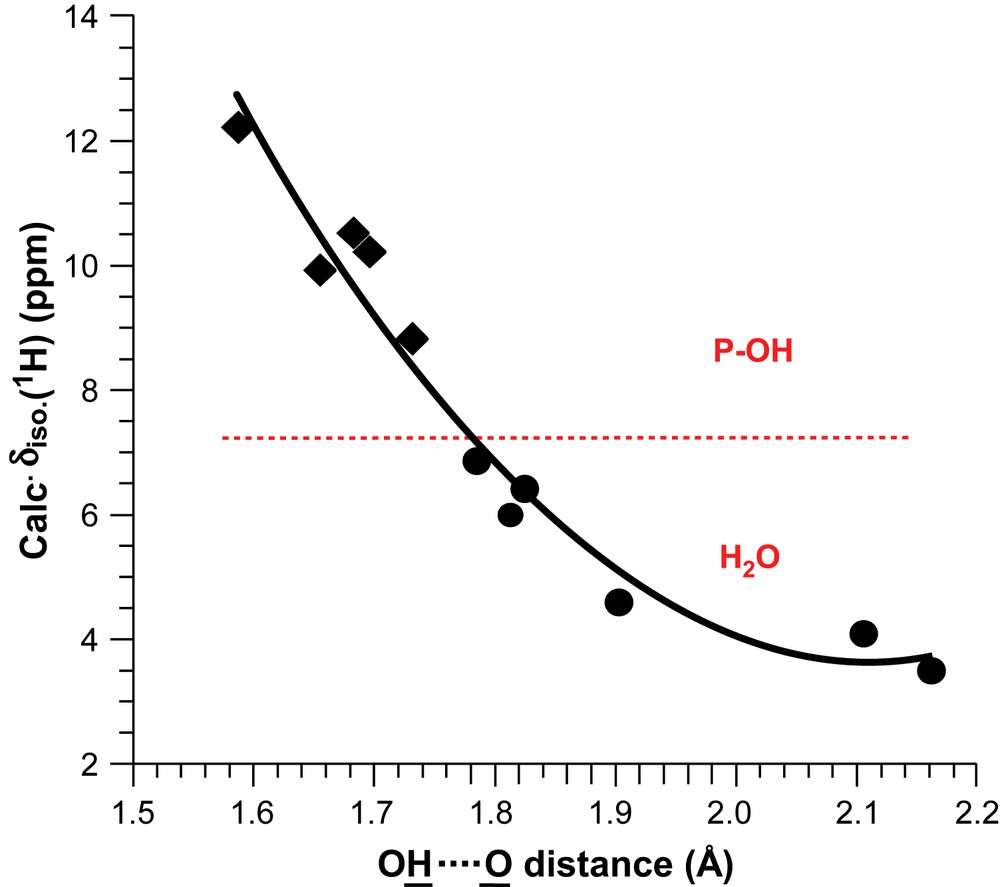
Correlation between the calculated 1H isotropic chemical shifts and the shortest internuclear distances for Brushite and MCPM.
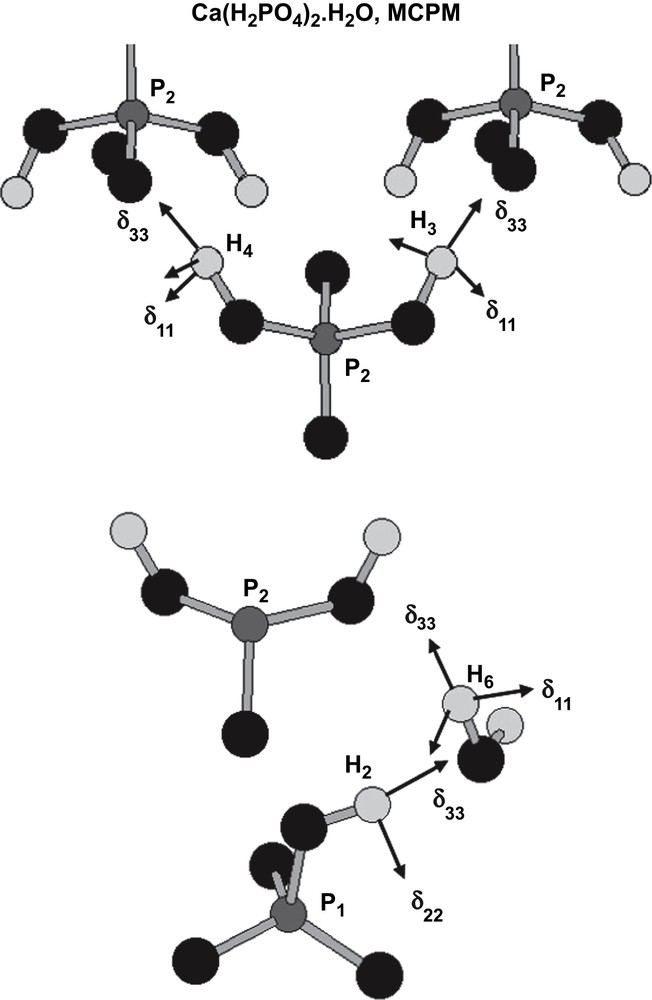
Absolute orientations of 1H tensors for MCPM (POH and H2O).
3.3 17O: calculated data
In the field of calcium phosphates, few 17O NMR results have been published until now to our knowledge. Only 17O MAS NMR spectra of Brushite and hydroxyapatite were previously reported [45].
The aim of the calculation of 17O NMR parameters in the various compounds presented in this paper is to determine whether there is a possibility to distinguish clearly the various types of oxygen environments present in calcium phosphates (Ca⋯O, P–O⋯Ca, P–O–P, P–OH and H2O) by using 17O NMR spectroscopy. The results are summarized in Table 1. The previously reported 17O NMR values for P–O–P sites in h-P2O5 [22], Ca⋯O in lime [46] (CaO) and P–O⋯Ca environments in brushite and HAp45 are in very good agreement with the ones obtained by the calculations. Moreover, it should be noticed that the relatively large 17O quadrupolar coupling constants calculated for water molecules in brushite and MCPM compare well with those experimentally observed in p-toluenesulphonic acid hydrate [47] and oxalic acid dihydrate [48]. Similarly, the 17O P–OH parameters are consistent with those measured in phenylphosphonic [49] and phenylphosphinic [42] acid.
Ca⋯O environments are expected in a very distinct area with high chemical shift values (around 300 ppm) and very small quadrupolar coupling constants (less than 1 MHz). Similarly H2O sites are well separated with small chemical shift values (≤0 ppm) and very large quadrupolar coupling constants (>7.5 MHz). On the other hand, P–O⋯Ca, P–O–P and P–OH environments appear in the same chemical shift range (between 65 and 115 ppm). To evaluate which type of oxygen environment can be clearly distinguished experimentally, we have calculated the 17O 3QMAS spectrum corresponding to the different oxygen sites (Fig. 7) [50]. Each 17O site gives rise to a sharp resonance in the δ3Qiso dimension at the position:
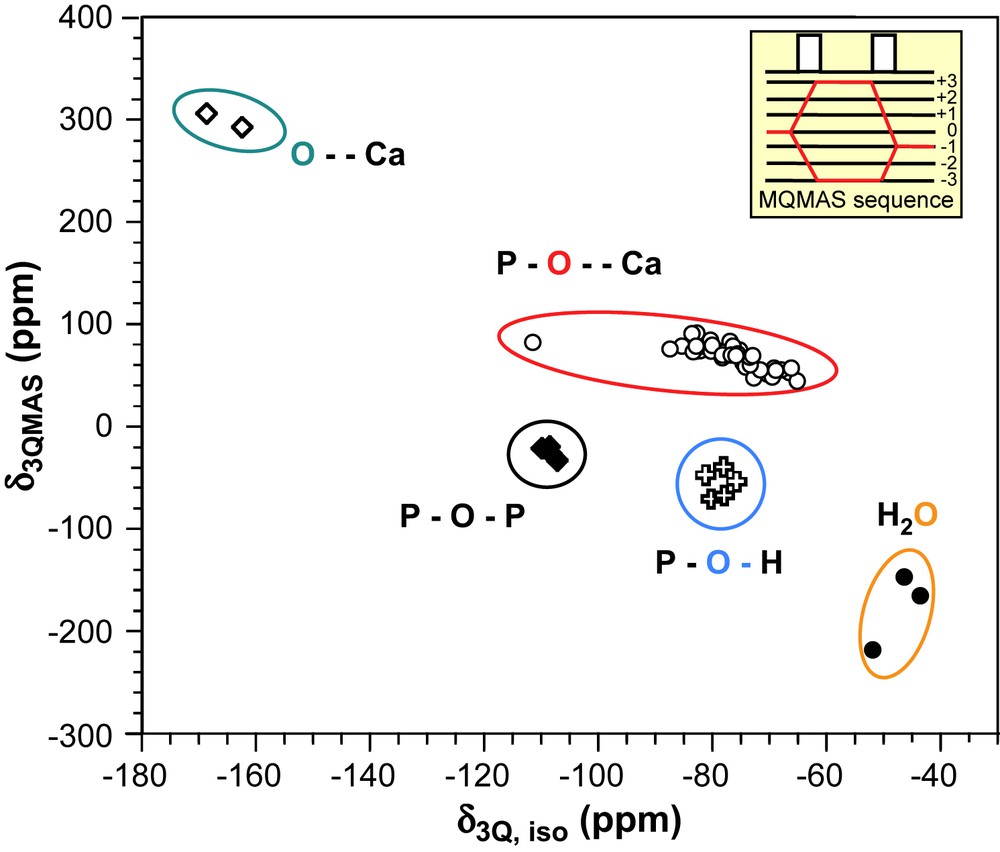
Theoretical positions of various 17O sites present in calcium phosphate phases in a 17O 3QMAS NMR experiment (B0 = 9.4 T).
The 2D spectrum shows that all types of oxygen environments investigated in this study appear in relatively distinct areas and should therefore be distinguishable using the 3QMAS NMR sequence. Indeed, even if P–O⋯Ca, P–O–P and P–OH environments have similar isotropic 17O chemical shifts, the discrimination is possible, thanks to different CQ values.
It can be noticed that the 17O chemical shift range observed for P–O⋯Ca environments is rather large and it was suggested that the shifts could discriminate between different P–O bond lengths in PO43− units [45]. Calculations show indeed that δiso (17O) tends to increase with P–O⋯Ca bond length.
4 Conclusions
It has been shown that first-principles calculations (using the GIPAW approach) are suitable for the interpretation of multinuclear solid-state NMR spectra of calcium phosphate phases, even in the case of non-fully resolved 1H MAS experiments. Full tensor data are obtained for 1H and 31P, including the absolute orientation of the principle axes. Interesting correlations between the δiso (1H) values and the shortest OH⋯O distances have been established, giving a more theoretical background to empirical data already published in the literature. Calculated 17O parameters tend to prove that the various oxygen sites can be separated in MQ-MAS experiments. Work is in progress in the laboratory for recording 17O spectra of enriched calcium phosphate phases.
Acknowledgments
Dr. D. Massiot (CRMHT) is warmly acknowledged for helpful discussions and for the access to the 750-MHz spectrometer in Orléans, France. The calculations have been performed in the IDRIS supercomputer centre of the CNRS (Project 51461).
Appendix Supporting information
The table includes fractional atomic coordinates of Ca4P2O9 structure after relaxation. The lattice parameters were not relaxed; a = 7.023 Å, b = 11.986 Å, c = 9.473 Å. The symmetry of the crystal was also constrained to be monoclinic P1211.
| Atom | x | y | z |
| Ca1 | 0.03135 | 0.34484 | 0.89371 |
| Ca2 | 0.53208 | 0.29963 | 0.89637 |
| Ca3 | 0.76779 | 0.30400 | 0.53484 |
| Ca4 | 0.26928 | 0.29958 | 0.25319 |
| Ca5 | 0.74235 | 0.10865 | 0.25339 |
| Ca6 | 0.25124 | 0.07842 | 0.01020 |
| Ca7 | 0.98997 | 0.06827 | 0.63431 |
| Ca8 | 0.51321 | 0.05225 | 0.58967 |
| O1 | 0.73015 | 0.51977 | 0.22758 |
| O2 | 0.60829 | 0.32156 | 0.28695 |
| O3 | 0.95391 | 0.36288 | 0.29568 |
| O4 | 0.77015 | 0.36447 | 0.05510 |
| O5 | 0.28916 | 0.41086 | 0.74965 |
| O6 | 0.09421 | 0.26407 | 0.62447 |
| O7 | 0.45747 | 0.24245 | 0.65781 |
| O8 | 0.32873 | 0.38221 | 0.48221 |
| O9 | 0.21655 | 0.00430 | 0.47360 |
| O10 | 0.07538 | 0.11672 | 0.26134 |
| O11 | 0.42032 | 0.10987 | 0.30057 |
| O12 | 0.24497 | 0.93066 | 0.22751 |
| O13 | 0.58489 | 0.10221 | 1.00048 |
| O14 | 0.83303 | 0.18754 | 0.84003 |
| O15 | 0.71748 | 0.99687 | 0.78546 |
| O16 | 0.92102 | 0.03030 | 0.01000 |
| O17 | 0.26471 | 0.26375 | 0.01636 |
| O18 | 0.76686 | 0.12650 | 0.48793 |
| P1 | 0.76573 | 0.39035 | 0.21539 |
| P2 | 0.28964 | 0.32559 | 0.62414 |
| P3 | 0.23669 | 0.04086 | 0.31668 |
| P4 | 0.76323 | 0.07675 | 0.91023 |