

1 Introduction
The chemistry of organic–inorganic hybrid materials is expanding because of the wide range of possibilities to access new kinds of nanomaterials. This field of research bridges different types of chemistry (organic, inorganic, and coordination) to materials science and also opens many possibilities for physical properties. The sol–gel methodology [1] represents a very efficient and powerful tool since it makes the materials science compatible with many different aspects of chemistry that were initially separated: solid-state chemistry, organic and inorganic chemistry, organometallic and coordination chemistry, macromolecular chemistry and also biomolecules [2–6]. All the molecular organic precursors containing at least two Si(OR)3 groups can be transformed into nanostructured hybrid materials by hydrolytic sol–gel polycondensation [7]. Thus a large variety of hybrid materials containing organic groups, chelating systems (cyclames, crown ethers, porphyrins…) and polymers have been obtained. All the systems studied until now are covalently bonded to the inorganic matrix and exhibit a self-organization at both the nanometric scale (X-ray) and the micrometric one (birefringence). These transformations are under kinetic control. They are strongly depending on the nature and the geometry of the organic units (linear, twisted, planar and even tetrahedral) [8–12]. The self-organization of organic units can be obtained by van der Waals interactions between organic groups [7]. A different organization can be obtained from H-bonding networks due to the presence of organic groups that favor the structuring of the hybrid solid [13,14].
Our current interest in the preparation of hybrid materials led us to explore the synthesis of nanostructured silica hybrids containing chiral organic units. Different methods can be used for inducing chirality in silicas and sol–gel materials [15]: the silylation of silica surfaces by silanes bearing a chiral group [16,17]; the use of chiral silanes as precursors in sol–gel polycondensation [18–20]; the composites obtained by physical entrapment of chiral molecules in silicas [21,22]; the imprinting with chiral templates [23,24]. The sol–gel process has been shown to be suitable for the immobilization of transition metal complexes and the derived materials are of interest in catalysis [25]. Enantioselective catalysis is a very interesting challenge in molecular chemistry. Asymmetric catalysis developed considerably during the last decades owing to its scientific importance. A huge number of chiral catalyst species have been described and used in asymmetric reactions such as asymmetric hydrogenations, epoxidations, and Diels–Alder reactions [26–28]. Tartaric acid has been used as chiral auxiliary in asymmetric catalysis because of its efficiency (i.e. sharpless epoxidation). Many composites of silica combined with optically active organic compounds have been prepared and used in chromatography and in heterogeneous catalysis [29,30]. Nowadays the design and synthesis of chiral hybrid materials with optical activity and enantioselectivity remain a challenge. We report in this work the synthesis of a new type of organic–inorganic hybrid materials containing chiral moieties prepared by sol–gel hydrolytic polycondensation of silylated-tartaramide derivatives.
2 Experimental
2.1 Materials and instruments
Reagent grade (R,R)-(+)-N,N,N′,N′-tetramethyl-l-tartaramide, (S,S)-(−)-N,N,N′,N′-tetramethyl-d-tartaramide, thallium ethoxide, allyl bromide and triethoxysilane (Aldrich) were used without purification. TBAF (1 M solution) and platinum divinyltetramethylsiloxane (3.5% platinum in poly(dimethylsiloxane), vinyl terminated) were purchased from Aldrich. All reactions were carried out under argon using a vacuum line and Schlenk techniques. Solvents were dried and distilled just before use.
Infrared spectra were recorded on an FTIR Perkin–Elmer 1600 neat for the precursors and in KBr pellets for the xerogels. 1H and 13C NMR spectra were performed on a Bruker AVANCE DPX 200 and on a Bruker WP-200 SY spectrometer (at 39.75 MHz for 29Si) operating on the Fourier transform mode using tetramethylsilane (TMS) as the internal standard and CDCl3 (Aldrich) as the solvent at room temperature. 29Si CP MAS NMR spectra were recorded on a Bruker FTAM 300 as were 13C CP MAS NMR spectra, in both cases, the repetition time was 10 and 5 s with contact times of 5 and 3 ms. Qn and Tn notations are given, respectively, for ((SiO)nSiO4−n) and ((SiO)n(R)SiO3−n) environments. The xerogel's density was measured on a Micromeritics AccuPyc 1330 using the helium pycnometry technique. The specific surface areas of the xerogels were obtained using a Micromeritics Gemini III 2375 after one-night vacuum (10−2 mbar) at 120 °C. X-ray diffraction experiments were carried out on solid powders in glass capillaries of 1-mm diameter in transmission configuration. A copper rotating anode X-ray source (4 kW) and 2D detector were used. The diffraction curves were obtained, giving the diffracted intensity as a function of the wave vector q. The diffracted intensity was corrected by exposure time, transmission and intensity background coming from diffusion by an empty capillary.
2.2 General procedure for the synthesis of bis-allyloxy-d- or l-tartaramide (2D or 2L)
To a solution of 5 g (24.5 mmol) of N,N,N′,N′-tetramethyl-tartaramide (1D or 1L) in toluene (200 mL) was added, under argon, 12.22 g (45.0 mmol) of thallium ethoxide (TlOEt) [31]. The mixture was left to react for 1 h. The solvent was evaporated and acetonitrile (150 mL) and allyl bromide 5.88 g (45.0 mmol) were added to the mixture. This solution was then stirred for 72 h, filtered on Celite and washed with CHCl3. The evaporation of solvent afforded 2,3-bis-triethoxysilylpropyl-N,N,N′,N′-tetramethyl-tartaramide (2D or 2L).
(R,R)-(+)-2,3-Bis-allyloxy-N,N,N′,N′-tetramethyl-l-tartaramide (2L): 6.26 g (90%); yellow oil, [α]D = +94.0° (c = 1, CHCl3). FTIR (neat, cm−1): ν 2933, 2859, 1646, 1502, 1403, 1258, 1136, 1079, 995, 926. 1H NMR (200 MHz, CDCl3): δ (ppm) = 5.99–5.85 (2H, m), 5.30 (2H, dd, J = 17.2 Hz and 1.6 Hz), 5.20 (2H, dd, J = 10.3 Hz and 1.3 Hz), 4.75 (2H, s), 4.16 (4H, dd, J = 7.0 Hz and 1.4 Hz), 3.16 (6H, s), 2.94 (6H, s). 13C NMR (50 MHz, CDCl3): δ (ppm) = 169.9 (CO), 134.9 (OCH2CHCH2), 117.6 (OCH2CHCH2), 77.4 (CHO), 71.5 (OCH2CHCH2), 37.7 (NCH3), 36.2 (NCH3).
(S,S)-(−)-2,3-Bis-allyloxy-N,N,N′,N′-tetramethyl-d-tartaramide (2D): 6.26 g (90%); yellow oil, [α]D = −94.0° (c = 1, CHCl3). FTIR (neat, cm−1): ν 2938, 2861, 1646, 1501, 1405, 1257, 1138, 1081, 995, 927. 1H NMR (200 MHz, CDCl3): δ (ppm) = 5.98–5.79 (2H, m), 5.26 (2H, dd, J = 17.2 Hz and 1.6 Hz), 5.15 (2H, dd, J = 10.5 Hz and 1.3 Hz), 4.71 (2H, s), 4.13 (4H, dd, J = 5.5 Hz and 1.2 Hz), 3.12 (6H, s), 2.89 (6H, s). 13C NMR (50 MHz, CDCl3): δ (ppm) = 169.8 (CO), 134.8 (OCH2CHCH2), 117.6 (OCH2CHCH2), 77.3 (CHO), 71.5 (OCH2CHCH2), 37.7 (NCH3), 36.2 (NCH3).
2.3 General procedure for the synthesis of bis-(alkoxysilyl)-d- or l-tartaramide (3D or 3L)
To 1.0 g (3.5 mmol) of 2,3-bis-allyloxy-N,N,N′,N′-tetramethyl-tartaramide (2D or 2L) were added, under argon, 1.85 g (11.0 mmol) of triethoxysilane and platinum divinyltetramethylsiloxane (3.5% platinum in poly(dimethylsiloxane), vinyl terminated). The mixture was left to react for three days. The evaporation of triethoxysilane excess afforded the respective 2,3-bis-triethoxysilylpropyl-N,N,N′,N′-tetramethyl-tartaramide (3D or 3L).
(R,R)-(+)-2,3-Bis-triethoxysilylpropyl-N,N,N′,N′-tetramethyl-l-tartaramide (3L): 1.28 g (60%); yellow oil, [α]D = +53.0° (c = 1, CHCl3). FTIR (neat, cm−1): ν 3481, 2973, 2928, 2885, 1650, 1399, 1080, 957, 792. 1H NMR (200 MHz, CDCl3): δ (ppm) = 4.77 (2H, s), 3.84 (12H, q), 3.56–3.49 (4H, m), 3.20 (6H, s), 2.95 (6H, s), 1.79–1.59 (4H, m), 1.25 (18H, t), 0.69–0.60 (4H, m). 13C NMR (50 MHz, CDCl3): δ (ppm) = 170.1 (CO), 78.1 (CHO), 72.5 (OCH2CH2CH2), 58.7 (OCH2CH3), 37.6 (NCH3), 36.1 (NCH3), 23.8 (OCH2CH2CH2), 18.5 (OCH2CH3), 6.9 (SiCH2CH2CH2). 29Si NMR (39.75 MHz, CDCl3): δ (ppm) = −45.0. Anal. Calcd for C26H56N2O10Si2: C, 50.95; H, 9.21; N, 4.57; Si, 9.16. Found: C, 51.65; H, 9.20; N, 5.63; Si, 7.07.
(S,S)-(−)-2,3-Bis-triethoxysilylpropyl-N,N,N′,N′-tetramethyl-d-tartaramide (3D): 1.5 g (70%); yellow oil, [α]D = −53.0° (c = 1, CHCl3). FTIR (neat, cm−1): ν 3502, 2973, 2928, 2883, 1650, 1398, 1080, 956, 792. 1H NMR (200 MHz, CDCl3): δ (ppm) = 4.72 (2H, s), 3.83 (12H, q), 3.61–3.52 (4H, m), 3.21 (6H, s), 2.95 (6H, s), 1.76–1.67 (4H, m), 1.25 (18H, t), 0.70–0.61 (4H, m). 13C NMR (50 MHz, CDCl3): δ (ppm) = 170.1 (CO), 77.9 (CHO), 72.5 (OCH2CH2CH2), 58.7 (OCH2CH3), 37.6 (NCH3), 36.1 (NCH3), 23.8 (OCH2CH2CH2), 18.6 (OCH2CH3), 6.9 (SiCH2CH2CH2). 29Si NMR (39.75 MHz, CDCl3): δ (ppm) −45.0.
2.4 General procedure for the synthesis of xerogels (X3D–X3L and X4D–X4L)
To a solution of 2,3-bis-triethoxysilylpropyl-N,N,N′,N′-tetramethyl-tartaramide (3D or 3L) (1.0 g, 1.6 mmol) and THF (817 μL) was added the catalyst TBAF (tetrabutylammonium fluoride) (2% mol of a 1 M solution) for X3D–X3L or HCl (2% mol) for X4D–X4L and water (90 μL, 3 equiv) and the mixture kept under static conditions at room temperature (25 °C). After ageing the obtained gel for seven days, it was powdered, washed with ethanol, acetone and ethyl ether and dried under vacuum (10−2 mbar) for 6 h to give pale yellow powders.
- (X3L): 0.312 g (50%). Tgel = 5 min. FTIR (KBr, cm−1): ν 3496, 2937, 2872, 1647, 1503, 1403, 1260, 1112, 788, 649. 13C CP MAS NMR: δ (ppm) = 170, 80–60, 37, 24, 10. 29Si CP MAS NMR: δ (ppm) = −59, −66, −100, −110. BET surface area < 10 m2 g−1. Density: 1.27 g cm−3. Anal. Calcd for C14H26N2O7Si2: C, 43.08; H, 6.66; N, 7.18. Found: C, 43.82; H, 7.58; N, 6.53.
- (X3D): 0.619 g (>99%). Tgel = 3 min. FTIR (KBr, cm−1): ν 3500, 2929, 2868, 1653, 1799, 1405, 1258, 1107, 788, 641. 13C CP MAS NMR: δ (ppm) = 170, 80–60, 36, 24, 10. 29Si CP MAS NMR: δ (ppm) = −59, −65, −100, −110. BET surface area < 10 m2 g−1. Density: 1.27 g cm−3. Anal. Calcd for C14H26N2O7Si2: C, 43.08; H, 6.66; N, 7.18. Found: C, 39.87; H, 7.35; N, 5.33.
- (X4L): 0.518 g (83%). Tgel = 90 min. FTIR (KBr, cm−1): ν 3494, 2928, 2867, 1646, 1512, 1421, 1266, 1117, 795, 659. 13C CP MAS NMR: δ (ppm) = 171, 80–60, 58, 36, 23, 19, 10. 29Si CP MAS NMR: δ (ppm) = −49, −58, −66, −93, −100. BET surface area < 10 m2 g−1. Density: 1.07 g cm−3. Anal. Calcd for C14H26N2O7Si2: C, 43.08; H, 6.66; N, 7.18. Found: C, 43.77; H, 7.45; N, 6.35.
- (X4D): 0.487 g (78%). Tgel = 84 min. FTIR (KBr, cm−1): ν 3503, 2988, 2855, 1650, 1782, 1408, 1233, 1100, 780, 646. 13C CP MAS NMR: δ (ppm) = 172, 80–60, 58, 35, 22, 19, 10. 29Si CP MAS NMR: δ (ppm) = −49, −58, −67, −93, −100. BET surface area < 10 m2 g−1. Density: 1.15 g cm−3. Anal. Calcd for C14H26N2O7Si2: C, 43.08; H, 6.66; N, 7.18. Found: C, 38.67; H, 6.23; N, 7.65.
3 Results and discussion
The new precursors were prepared from the two enantiomers (R,R)-(+)-N,N,N′,N′-tetramethyl-l-tartaramide (1L) and (S,S)-(−)-N,N,N′,N′-tetramethyl-d-tartaramide (1D) in two steps as outlined in Fig. 1.
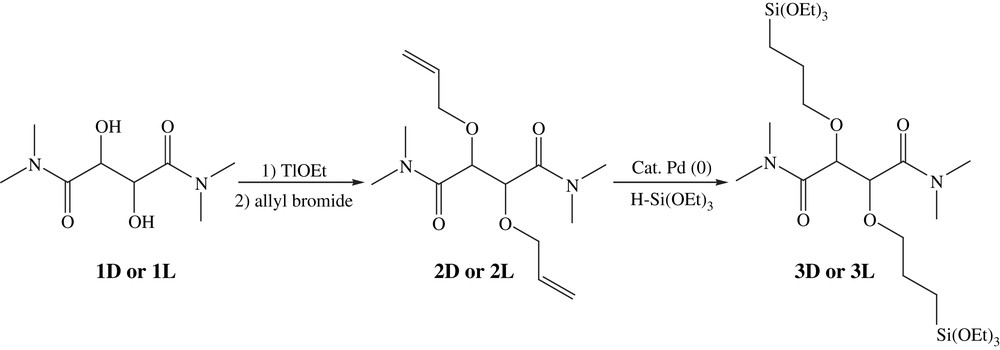
Preparation of derivatives 2D–2L and 3D–3L.
The first step consisted of the formation of thallium alkoxide [31] followed by the etherification with allyl bromide. The second was the hydrosilylation of diallyl derivative (2D–2L) with triethoxysilane in the presence of Karstedt catalyst [32]. The hydrolysis–polycondensation of the resulting alkoxy precursors (3D–3L) allowed the formation of chiral bridged polysilsesquioxanes (X3D–X3L using TBAF as catalyst and X4D–X4L with HCl) (Fig. 2).
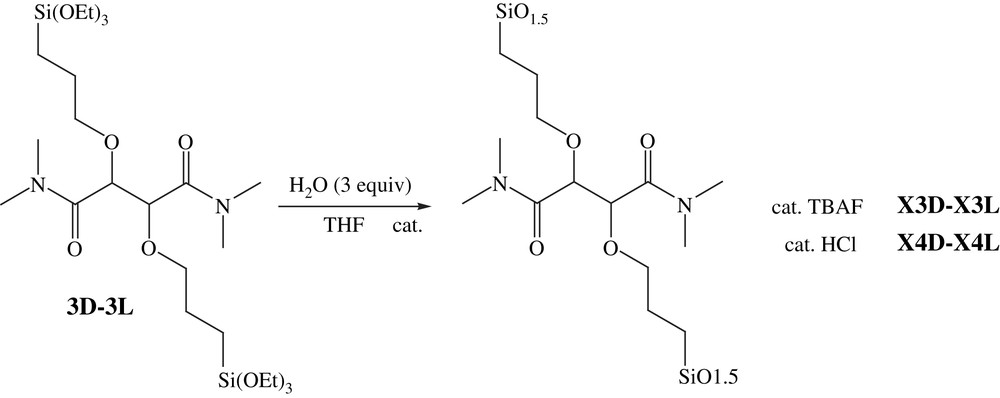
Preparation of xerogels X3D–X3L and X4D–X4L.
The gels were aged for seven days, and then treated as usual to give the xerogels, which were analysed by BET, NMR and X-ray diffraction techniques. Non-porous solids were obtained in all the cases (SBET < 10 m2 g−1).
13C CP MAS NMR spectra of X3D–X3L and X4D–X4L revealed the presence of the organic units in the inorganic network. The signals that attributed to the organic core were centred at 10 ppm (OCH2CH2CH2Si), 22–24 ppm (OCH2CH2CH2Si), 35–37 ppm (NCH3), 60–80 ppm (OCH2CH2CH2Si) and (CHOCH2CH2CH2), 170–172 ppm (CO). In the case of HCl catalysis some remaining triethoxysilyl groups appeared at 19 ppm (SiOCH2CH3), and 58 ppm (SiOCH2CH3), in agreement with the level of condensation at silicon deduced from the 29Si CP MAS NMR spectra. This level was estimated in first approximation by deconvolution of the spectra. It has been previously shown that no significant variation in relative peak intensity was observed in the case of such alkylene or arylene hybrid solids by comparison between CP MAS and single-pulse experiments that allow a quantitative evaluation [33,34]. Moreover, since materials of the same precursor are compared, it can be assumed that the relative peak intensity can be used as an indicator of the order of magnitude for the level of condensation, which was calculated according to the general equation: L.C. = [0.5(T1 area) + 1.0(T2 area) + 1.5(T3 area)]/1.5. The spectra indicated, for all samples, that the obtained xerogels were well condensed. The materials obtained using HCl as the catalyst presented a lower degree of condensation compared to TBAF. The degree of polycondensation, using TBAF as a catalyst, was 96% for X3D and 97% for X3L, with T2 [C–Si(OR)(OSi)2] signal at around −59 ppm and T3 [C–Si(OSi)3] signal at −66 ppm as the major peak. Using HCl as catalyst, degrees of condensation of 73% and 76% were obtained for X4D and X4L, respectively, with T1 located at around −49 ppm, T2 at −58 ppm and T3 at −66 ppm.
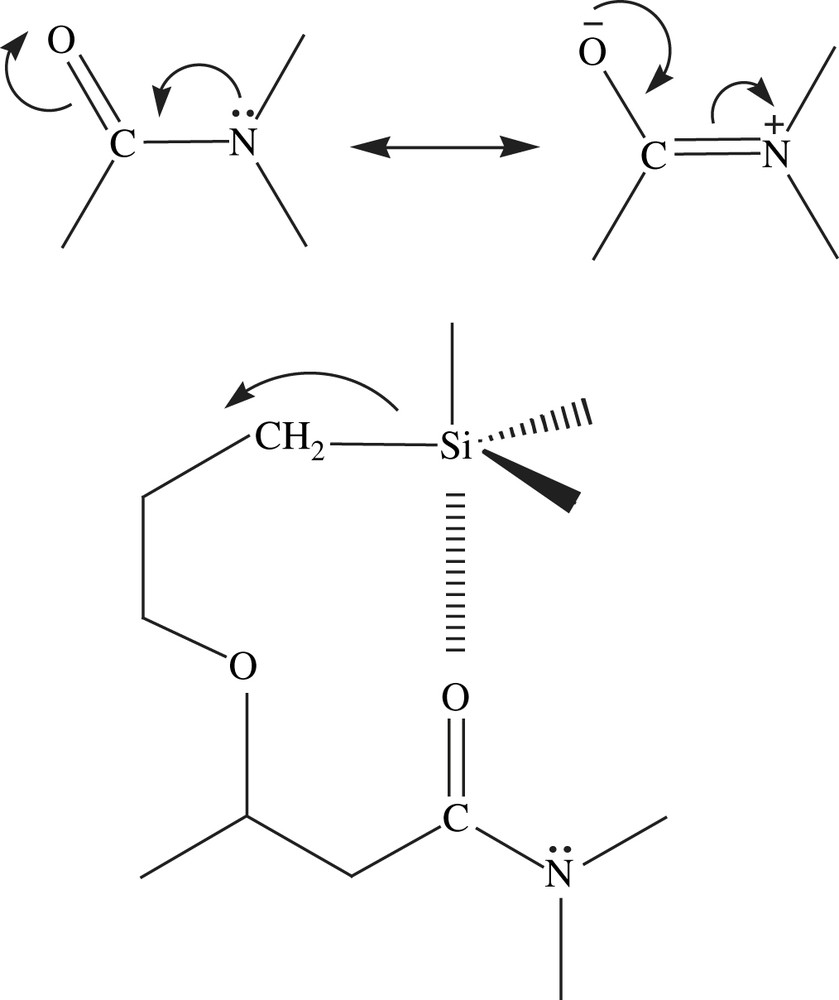
Interestingly, additional weak signals located at −100, −110 ppm (TBAF) and −93, −100 ppm (HCl), corresponded to Q substructures, which showed that a few cleavage of Si–C bond occurred during the sol–gel process. It is worth to mention that such a cleavage was never observed before during the polycondensation of nanostructured materials [7]. The explanation is, certainly, the intramolecular activation of the carbonyl group by the lone pair of the nitrogen atom that could induce the silicon–carbon bond breaking (Scheme 1). The nucleophilic activation of the nucleophilic substitution at silicon is a well-known process.[35]
X-ray powder diffraction patterns of these materials are given in Fig. 3. Like other nanostructured hybrid materials, they did not exhibit narrow Bragg peaks. For each sample, the presence of broad reflexions only ruled out the occurrence of long-range order in the condensed state. However, as a first approximation assuming Bragg's law a priori, the distances associated with the q values were determined (d = 2π/q) and are given in Table 1. The nature of the catalyst used for the hydrolysis–polycondensation reaction appeared to have a significant influence on the organization at the nanometric scale since the X-ray powder diffraction (XRD) patterns of the xerogels X3D–X3L and X4D–X4L were different (Fig. 3).
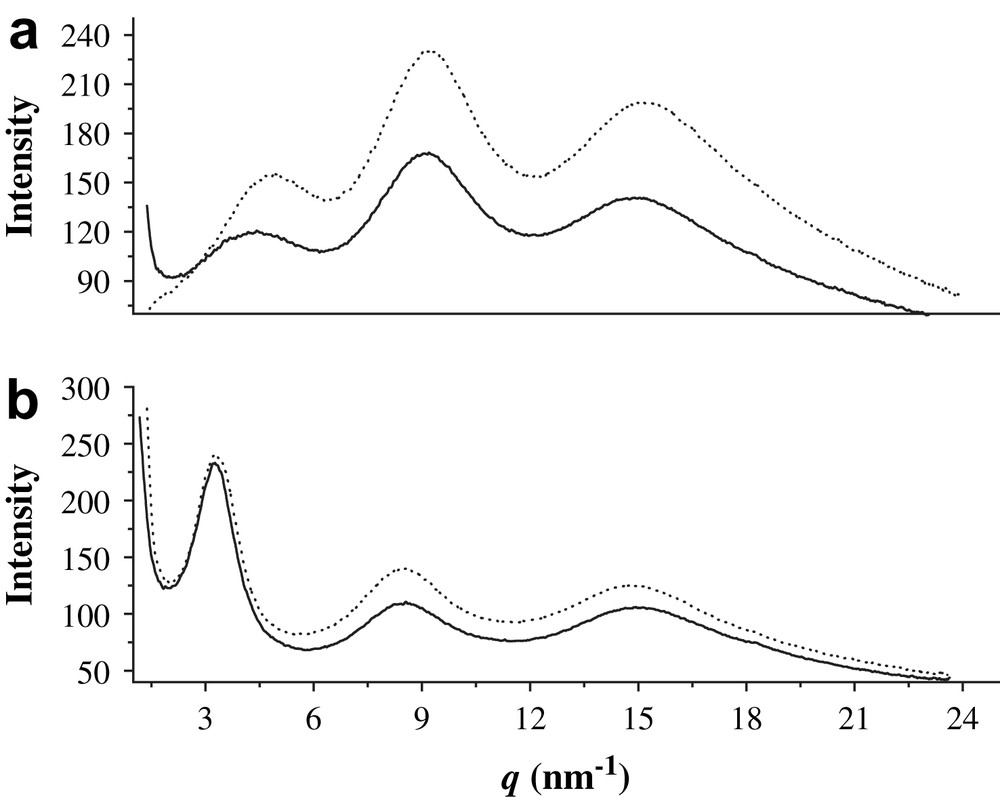
XRD patterns of xerogels: (a) X3D (solid line) and X3L (dotted line) and (b) X4D (solid line) and X4L (dotted line).
X-ray powder diffraction (XRD) patterns of chiral xerogels
| Xerogels | d (nm) |
| X3D | 1.92/0.75/0.42 |
| X3L | 1.92/0.74/0.42 |
| X4D | 1.46/0.69/0.42 |
| X4L | 1.27/0.68/0.41 |
In all the cases, a broad signal centred at 15 nm−1 (0.42 nm) was present, as already observed in all other nanostructured silica-based hybrid materials, and was attributed to the contribution of Si–O–Si units. The organization appeared to be dependent on the catalyst employed.
4 Conclusion
We report here the synthesis of new chiral hybrid organic–inorganic materials containing tartaramide units. The chiral units are unambiguously inserted inside the materials. Work is in progress in order to test their ability as support for enantiomeric separations and also for homogeneous catalysis.
Acknowledgements
The authors thank Dr Philippe Dieudonné (LCVN – UMR 5587, Montpellier, France) for XRD measurements, Dr Damien Hérault for a partial contribution, the CNRS and the “Université Montpellier-2” for financial support and CAPES and MENERS for Post-Doc fellowships (Brazil).