

1 Introduction
The versatility of the naturally occurring terpenes and especially of the readily available pinenes furnishes the starting material to access chiral auxiliaries which have found a variety of applications in asymmetric catalysis. Especially, N,N [1–4], P,N [3,5–7], P,P [8] and P,O [9] type chiral ligands have been prepared and applied in various enantioselective processes such as allylic alkylation [2,9], allylic oxidation [2,4], cyclopropanation [1,2], reduction of ketones [3], Heck reaction [5] and hydrogenation [6–8]. On the other hand, the pinene framework has also been utilized to synthesize chiral aminoalcohols which found use, via oxazaborolidines in reduction of CO [10–12] when associated to aluminium in Diels–Alder reactions [12] and finally in addition of diethylzinc to aldehydes [13].
On the other side, asymmetric catalytic transfer hydrogenation [14] using isopropanol as the hydrogen source has been developed extensively during these last few years because it is an easy method to apply and very efficient for the reduction of prochiral substrates into optically active products without using hydrogen gas. A large variety of catalysts has been applied including ruthenium [15], rhodium [16] and iridium [17] associated with chiral ligands.
Among the ligands tested and following the work of Noyori in this field, essentially two successful series have been developed, one based on 1,2-aminoalcohols [18] and the second one on monotosyl diamines [14,19], both coordinated to arene ruthenium type catalysts. Investigations on the origin of the enantioselectivity with these ligands support a ligand cooperativity based mechanism with a concerted delivery of a proton from a NH of the ligand and a hydride from ruthenium [20,21]. Within this context, the development of new chiral ligands remains an interesting challenge. In line with our ongoing interest in the synthesis of chiral ligands from the chiral pool [22], we have been interested in using chiral terpenic starting compounds to prepare new bifunctional ligands bearing a NH and a OH functionality, the latter having been found beneficial to the transfer hydrogenation process assisted by ruthenium catalysts. In this context, α-pinene has been chosen as the starting material. Here, we report on the synthesis of new β-aminoalcohol, diiminoalcohol, and diaminoalcohol ligands with an α-pinene framework and their evaluation in transfer hydrogenation of acetophenone.
2 Results and discussion
2.1 Synthesis
Two methods have been reported for the synthesis of β-aminoalcohols based on α-pinene bearing a NH2 moiety. The first one involves an osmium-induced vicinal oxyamination of α-pinene in the presence of the imidoosmium derivative tBuNOsO3 and uses stoechiometric amounts of osmium [23]. We applied the second oxidation method which is depicted in the Fig. 1 [24]. Thus, (-)-α-pinene 1 (81% ee optical purity) was first oxidized with potassium permanganate into the (1R,2R,5R)-hydroxyketone 2. This reaction is highly stereoselective and the oxidant reacts on the side opposite to the gem-dimethyl bridge [25]. Compound 2 was then reacted with 50% aqueous hydroxylamine yielding the corresponding oxime 3. This compound was recrystallized from diethylether-hexane providing the optically pure oxime 3, as assessed by Masui and Shioiri [24a]. Finally, the reduction of 3 with lithium tetrahydridoaluminate afforded the β-aminoalcohol 4.HCl. The β-aminoalcohol 4 was obtained after neutralization with sodium hydroxide (Fig. 1).
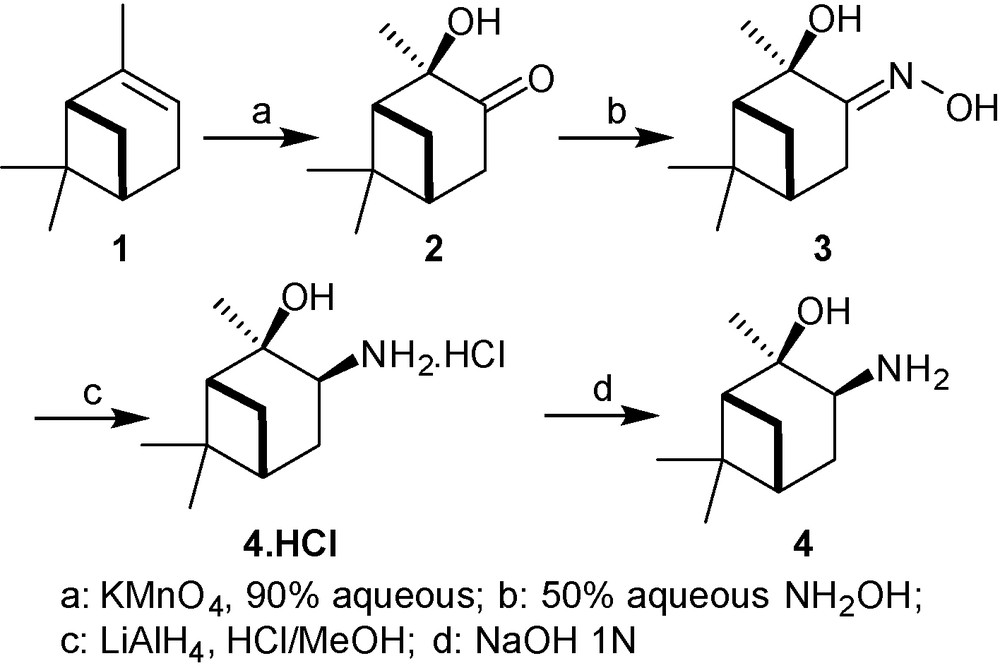
Synthesis of the β-aminoalcohol 4 from α-pinene.
Next, the substitution of the amino moiety of 4 by different groups has been performed according to three procedures as depicted in the Fig. 2. Benzyl and 4-phenylbenzyl moieties have been introduced by reaction of the hydrochloride salt 4.HCl with the selected aldehyde. Then, reduction of the intermediate imines by NaBH4 led respectively to the crude 3-(benzylamino)-2-hydroxypinane 5 and 3-(biphenyl-4-ylmethylamino)-2-hydroxypinane 6. These compounds were recrystallized from diethylether-petroleum ether mixtures affording pure 5 and 6 in 63 and 68% yield, respectively. Methylation of the amine 4 has been realized in the presence of 1,1’-carbonyldiimidazole [24]. After reduction with LiAlH4 of the intermediate urea and recrystallization from hexane, N-methyl aminoalcohol 7 was obtained in 61% yield. Tosylation of 4 could be carried out classically in the presence of paratoluene-sulfonylchloride providing 8 in 92% yield [24]. The new ligands 5 and 6 have been fully characterized (Fig. 2).
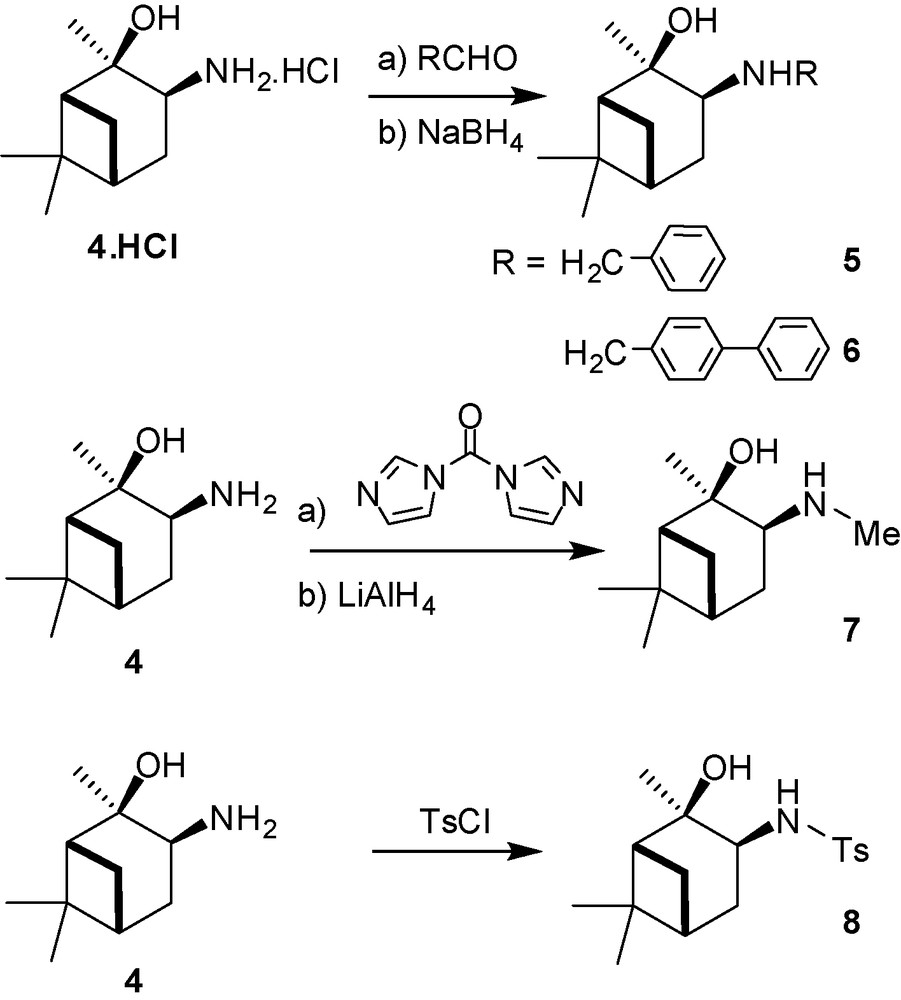
Synthesis of the N-substituted aminoalcohol ligands 5–8.
We then turned our attention to the preparation of C2-symmetric diaminoalcohol derivatives. First, the diiminodiols 9 and 10 could be easily synthesized by reaction of two equivalents of the hydroxyketone 2 with an equivalent of a diamine in the presence of APTS and elimination of water in a Dean–Stark apparatus. These diiminodiols 9 and 10 could then be reduced into the corresponding diamines by NaBH4. Thus, we used this procedure with 1,2-diaminoethane and 1,3-diaminopropane which provided the diimines 9 and 10 in 68 and 51% yield, respectively, and the diamines 11 and 12 in, respectively, 40 and 65% yield from the imines. As reported in the literature, the reduction appeared to be stereoselective and no formation of diastereoisomers was observed by NMR analysis [26]. Thus, in agreement with the formation of the aminoalcohol 4, we propose the same stereoselectivity for ligands 11 and 12. Ligands 9–12 have been fully characterized (Fig. 3).
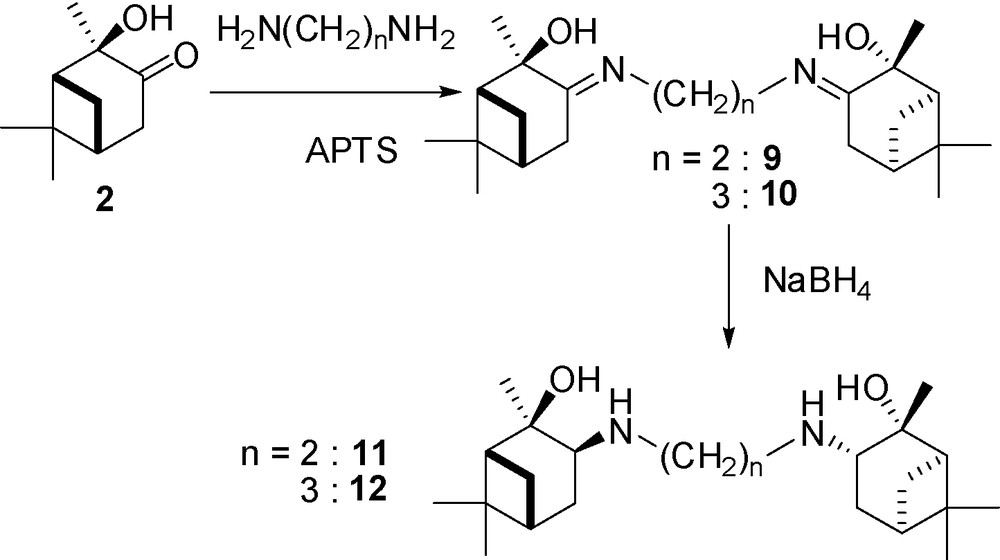
Synthesis of diimino- and diamino-diol ligands 9–12.
2.2 Evaluation of ligands 3–12 in the ruthenium-catalyzed transfer hydrogenation of acetophenone
Next chiral derivatives 3–12 were applied in the ruthenium catalyzed transfer hydrogenation of the standard substrate acetophenone (Fig. 4). The catalysts were generated by mixing the [RuCl2(p-cymene)2]2 precursor with the desired ligand in the presence of KOH. The results are summarized in Table 1.
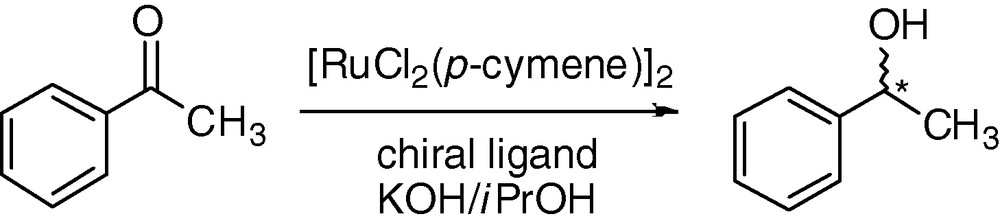
Asymmetric transfer hydrogenation of acetophenone.
Transfer hydrogenation of acetophenone in the presence of [RuCl2(p-cymene)2]2 as catalytic precursora.
| Entry | Ligand | T (°C) | Conversion (%)b | ee (%)c |
| 1 | 3 | 20 | 71 | 8 |
| 2 | 4 | 20 | 85 | 18 |
| 3 | 5 | 20 | 83 | 34 |
| 4 | 6 | 20 | 77 | 45 |
| 5 | 7 | 20 | 92 | 29 |
| 6 | 8 | 20 | – | – |
| 7 | 9 | 80 | 90 | 10 |
| 8 | 10 | 80 | 97 | 18 |
| 9 | 11 | 80 | 90 | 13 |
| 10 | 12 | 80 | 88 | 7 |
a All reactions were performed during 3 h by using 2 mmol of acetophenone in 20 mL iPrOH. S/Ru = 100, Ligand/Ru = 2, KOH = 0.1 mmol.
b Determined by GC analysis on a CP-Sil 5 CB capillary column.
c Determined by GC on a Chirasil–Dex capillary column. The configurations of the alcohol were always (R).
As is shown in the table, the hydrogenation of acetophenone can be achieved by Ru(II) complexes modified by ligands 3–12. In the case of the β-hydroxyoxime 3, the transfer hydrogenation occurred in 71% conversion after 3 h at room temperature with a low ee of 8%. In the presence of ligand 4 containing a primary amine, under identical catalytic conditions, a slightly higher conversion was achieved (85%) but the selectivity remained low (18% ee). These results show, as expected, that the presence of an NH moiety is preferred to have an efficient ligand cooperation during the transfer of the hydrogen atoms to the substrate. The reactivity obtained in the case of ligand 3 could perhaps be due to a partial in situ reduction of the oxime ligand 3 into the amino 4 compound but up to now, this assumption could not be proved.
The selectivity of the hydrogenation could be enhanced by substitution of the NH2 group of 4 by a methyl (7, 29% ee), a benzyl (5, 34% ee), and best a 4-phenylbenzyl moiety (6, 45% ee). This selectivity increase correlates with the steric hindrance of the nitrogen substituents. The influence of the nature of the substituent on the amino group had been systematically screened using (1S,2R)-norephedrine as aminoalcohol: the best result in the reduction of ethylacetocetate in terms of both enantioselectivity and reactivity was also obtained by using a 4-phenylbenzyl substituted amine [27].
In the case of ligand 8 bearing a tosyl moiety on the nitrogen atom, no reaction occurred at all. An identical observation was made by our group when we applied a tosyl-substituted ephedrine modified ruthenium complex in the hydrogenation of ethylacetoacetate [27]. In our hands, the same observation was made using acetophenone as substrate and tosylated (S)-alaninol or (R)-leucinol as ligand [28]. In the presence of KOH, the double deprotonation of the N-tosyl hydroxy ligand 8 can provide, upon reaction with the ruthenium precursor, an amido-alcoxo-ruthenium complex which is not providing any active catalyst in the presence of isopropanol.
In the presence of the chiral diimine and diamine auxiliaries 9–12, a reaction temperature of 80 °C was necessary to reduce acetophenone. In that case, conversions between 88 and 97% could be reached even with the diimines 9 and 10. Alper has previously reported on the hydrogenation of acetophenone catalyzed by a ruthenium complex associated with a chiral Schiff base derived from diaminocyclohexane. Similar conversions with moderate ee's (8–28%) were obtained. In our case, the enantioselectivities lie within the same range for the two diamines as the ee's did not exceed 18% [29].
3 Conclusion
In summary, optically pure β-aminoalcohols, diimino, and diaminalcohols are conveniently prepared from α-pinene in a few steps and in moderate to good overall yields. The use of these ligands in transfer hydrogenation allowed the reduction of acetophenone with good activities and moderate enantioselectivities. Work is under progress to apply these ligands in other asymmetric processes. As aminoalcohols have proved to be the most selective within this series of bifunctional ligands, further studies are also in progress to synthesize new aminoalcohols from α-pinene and other optically pure terpenes and to apply all new ligands in asymmetric processes.
4 Experimental section
All the reactions were carried out under nitrogen atmosphere using standard Schlenk techniques. 1H and 13C NMR spectra were recorded on Varian Gemini – 300 MHz and Varian Innova – 400 MHz spectrometers. Optical rotations were measured on an ZUZ: Modelo 412 polarimeter. Elemental analysis was performed by “Service central d’analyse du CNRS”.
The hydroxyketone 4 was prepared according to reported protocols [24,30].
4.1 Synthesis of the ligands
4.1.1 (1R,2R,3S,5R)-3-(benzylamino)-2,6,6-trimethyl-bicyclo[3.1.1]heptan-2-ol 5
A mixture of aminoalcohol hydrochloride 4.HCl (1.37 g, 6.7 mmol), benzaldehyde (0.711 g, 6.7 mmol) and triethylamine (0.77 g, 1.05 mmol) in 15 ml ethanol was stirred at room temperature for 30 min. NaBH4 (0.45 g, 12 mmol) was then added dropwise at 0 °C and the resulting solution was stirred for 20 min at room temperature. The reaction mixture was neutralized with water (2.5 ml) and CH2Cl2 (10 ml) was added. The solution was filtered and solvents were evaporated under reduced pressure. The remaining residue was extracted with diethylether (5 ml), washed with water (3 10 ml) and dried over Na2SO4. The solvents were removed under reduced pressure and the product was purified by recrystallization from diethylether/ petroleum ether to afford the N-benzylamino-alcohol 5.
Yield: 63%. [α]D20 = −14.8 (c 1.0, CHCl3). 1H NMR (CDCl3) δ 0.91(s, CH3), 0.98 (s, CH3), 1.25 (s, CH3), 1.5–1.7 (m, 6H), 2.63 (m, CHN), 3.8 (dd, CH2), 7.1–7.5 (m, Harom); 13C NMR (CDCl3) 20.1; 22.5; 25.56; 27.05; 28.21; 39.8; 41.22; 50.61; 52.63; 67.15; 75.55; 126.56; 127.61; 127.77; 139.75. Anal. calculated for C17H25NO: C, 78.72; H, 9.71; N: 5.04. Found: C, 78.45; H, 9.60; N, 4.95.
4.1.2 (1R,2R,3S,5R)-3-(biphenyl-4-ylmethylamino)-2,6,6-trimethylbicyclo[3.1.1]heptan-2-ol 6
The same procedure was used with biphenylcarbaldehyde as precursor to afford aminoalcohol 6.
Yield: 68%. [α]D20 = −18.7 (c 1.0, CHCl3). 1H NMR (CDCl3) δ 0.91(s, CH3), 0.98 (s, CH3), 1.25 (s, CH3), 1.5–1.7 (m, 6H), 2.63 (m, CH-N), 3.8 (dd, CH2), 7.1–7.8 (m, Harom); 13C NMR (CDCl3) 20.1; 22.5; 25.56; 27.05; 28.21; 39.8; 41.22; 50.61; 52.63; 67.15; 75.55; 120.51; 127.39; 133.0; 133.26; 135.7; 142.11; 143.27; 144.31. Anal. calculated for C23H29NO: C, 82.34; H, 8.71; N, 4.18. Found: C, 82.11; H, 8.56; N, 4.02.
4.1.3 (1R,1’R,2R,2’R,5R,5’R)-3,3’-(ethane-1,2-diylbis-(azan-1-yl-1-ylidene))bis(2,6,6-trimethylbicyclo-[3.1.1]heptan-2-ol) 9
A mixture of (1R,2R,5R)-hydroxyketone 2 (5 g, 0.0297 mol), ethane-1,2-diamine (0.894 g, 0.0148 mol) and a few crystals of APTS in 50 mL of toluene was refluxed for 24 h in a Dean–Stark apparatus. The resulting mixture was filtered, the filtrate was concentrated under reduced pressure to a 5 mL volume and 20 mL of ethanol were added. The resulting solution was cooled to 0 °C and water was added to crystallize the diiminodiol. The precipitate was filtered and washed with hexane to give 1.7 g of the diiminodiol 9.
Yield: 68%. [α]D20 = −13.1 (c 1.0, CH3OH). 1H NMR (CDCl3) δ 0.88 (s, 6H), 0.98 (s, 6H), 1.25 (s, 6H), 1.56 (d, 2H), 2.05 (m, 4H), 2.36 (m, 4H), 3.65 (m, 4H), 13C NMR (CDCl3) 23.08, 25.35, 27.47, 28.27, 28.43, 33.78, 38.55, 50.53, 51.57, 67.99, 177.14 (CN).
4.1.4 (1R,1’R,2R,2’R,5R,5’R)-3,3’-(propane-1,3-diylbis-(azan-1-yl-1-ylidene))bis(2,6,6-trimethylbicyclo[3.1.1]-heptan-2-ol) 10
The same procedure was applied using 1,3-diaminopropane as precursor.
Yield: 40%. [α]D20 = −6.3 (c 1.0, CH3OH). 1H NMR (CDCl3) δ 0.85 (s, 6H), 1.32 (s, 6H), 1.47 (s, 6H), 1.52 (d, 2H), 1.67 (d, 4H), 1,9 (m, 2H), 2.06 (m, 2H), 2.36 (m, 2H), 2.51–2.64 (m, 4H), 3.39 (t, 2H), 13C NMR (CDCl3) 23.02, 27.47, 28.28, 28.49, 31.26, 33.43, 38.50, 48.37, 50.50, 52.34, 68.28, 177.17.
4.1.5 (1R,1’R,2R,2’R,3S,3'S,5R,5’R)-3,3’-(ethane-1,2-diylbis(azanediyl))bis(2,6,6-trimethylbicyclo[3.1.1]-heptan-2-ol) 11
The diiminodiol 9 (0.5 g, 1.3 mmol) dissolved in 15 mL absolute ethanol was cooled to 0 °C. Sodium tetraborohydride (0.11 g, 2.9 mmol) was then added in small portions. The solution was stirred 30 min at this temperature then quenched with water (2.5 mL) and concentrated under reduced pressure. The residue was extracted with diethylether (3 × 30 mL) and dried over MgSO4. Evaporation of the solvent provided 0.52 g of the diamine 11.
Yield: 51%. [α]D20 = +8.3 (c 1.0, CH3OH). 1H NMR (CDCl3) δ 0.87 (s, 6H), 0.96 (s, 6H), 1.31 (s, 6H), 1.35–1.75 (m, 12H), 2.64 (m, 2H), 2.67 (m, 4H), 13C NMR (CDCl3): 22.84, 23.05, 24.71, 27.28, 28.11, 39.01, 40.21, 50.36, 50.85, 60.89, 77.63. Anal calculated for C22H40N2O2: C, 72.48; H, 11.06; N,7.68. Found: C, 72.64; H, 11.22; N, 7.51.
4.1.6 (1R,1’R,2R,2’R,3S,3'S,5R,5’R)-3,3’-(propane-1,3-diylbis(azanediyl))bis(2,6,6-trimethylbicyclo[3.1.1]-heptan-2-ol) 12
The same procedure was applied starting from the diimine 10.
Yield: 65%. [α]D20 = −22.7 (c 1.0, CH3OH). 1H NMR (CDCl3) δ 0.82 (s, 6H), 1.22, (s, 6H), 1.34 (s, 6H), 1.41–1.52 (m, 12H), 1.55 (m, 2H), 2.55 (m, 4H), 2.64 (m, 2H), 13C NMR (CDCl3): 21.55, 22.47, 24.95, 26.14, 27.06, 28.16, 39.86, 41.11, 46.09, 50.57, 68.03, 75.67. Anal calculated for C23H42N2O2: C, 72.97; H, 11.18; N, 7.40; O, 8.45. Found: C, 73.12; H, 11.25; N, 7.22.
4.2 Catalytic experiments
The catalysts were generated in situ prior to catalysis by heating a mixture of the [RuCl2(p-cymene)2]2 complex with the desired aminoalcohol at 80 °C for 20 min in dry propan-2-ol. Then, a solution of the substrate in propan-2-ol followed by KOH was added to the mixture. Conversions were determined by CG analysis on a CP-Sil 5 CB column and the enantiomeric excesses were calculated from chiral GC analysis with a Chirasil–Dex capillary column. Absolute configuration was assigned by comparing with an authentic sample following Noyori procedure [17e].
Acknowledgments
We are grateful to the “ministère des Affaires étrangères (programme Volubilis)”, the CNRS and the “ministère de l’Éducation nationale, de l’Enseignement supérieur et de la Recherche scientifique du Maroc” for financial support.