

1 Introduction
Conventional solvents are usually volatile organic compounds (VOCs), which are harmful for the environment and human health. VOCs that spill into the environment contribute to tropospheric ozone formation and contaminate water and soil.
Accordingly, new regulations introduced in the 1990s (European directives 1999/13/EC, 2001/81/EC, 2004/73/EC, REACH, Clean Air Act) have forced the industry to find solutions to reduce their emissions and waste released into the environment. One potential solution involves the replacement of conventional solvents by new alternative solvents, serving as a preventive measure.
Biosolvents, based on natural ingredients, have been developed to offer the advantages required for an alternative to fossil resources. Such molecules are derived from vegetable, animal or mineral raw materials, using chemical and physical processes that are safer for the environment and humans. The natural synthon must represent a major part of the final product. Moreover, because their conception is compatible with sustainable development, biosolvents are expected to provide a positive environmental balance (VOC reduction, biodegradability, non ecotoxicity) and to improve safety aspects.
The major classes of biosolvents are esters of natural organic acids (produced by fermentation of renewable materials), fatty acid esters, bioethanol, terpenic compounds, isosorbide and glycerol derivatives [1,2].
These biosolvents are incorporated into different formulations for cleaning applications [3,4], cosmetics [5,6], inks [7], paintings [8], bituminous flux [9,10] and agricultural chemicals [11,12].
In this work, new biosolvents were developed to substitute the petrochemical solvents used in nail polish formulations.
To identify these alternative biosolvents, several criteria have been taken into account (Table 1):
- • technical specifications: the biosolvent has to display the appropriate levels of performance and security required for the considered application. The primary role of a solvent is to dissolve all the components of the formulation giving a homogeneous solution. Appropriate volatility of the molecules is essential to limit the drying time of the formulation. However, a compromise must be found to provide the optimal level of volatility, keeping VOC emissions to a minimum. Moreover, the flash point has to be high enough during the production, conditioning and transportation phases for security purposes;
- • environmental and sanitary criteria have to be met to satisfy the specific regulatory, standards and ecolabel specifications;
- • the processes involved in solvent production have to be eco-compatible: they have to respect most of the 12 principles of green chemistry proposed by Anastas and Warner;
- • the selection of renewable raw materials depends on their availability, their cropping cycle, their physicochemical properties and their cost.
Criteria for the selection of new solvents.
| Criteria | Measurements | Specifications |
| Solvent properties | Solubility parameters | Technical |
| Kauri index | ||
| Toxicity/ecotoxicity/biodegradability | Solvent properties | Sanitary/environmental |
| Volatility | Vapor pressure | Technical/environmental |
| Boiling point | ||
| Evaporation index | ||
| Flammability | Flash point | Technical/safety |
| Raw materials | Origin | Environmental/economic |
| Availability | ||
| Cost | ||
| Chemical process | Green chemistry indicators | Environmental/economic |
| Cost |
To meet the major criteria, we have targeted different molecules capable to comply with the technical specifications such as nitrocellulose solubilisation. We observed that the performing molecules represented in Fig. 1 could be obtained from short alcohols.
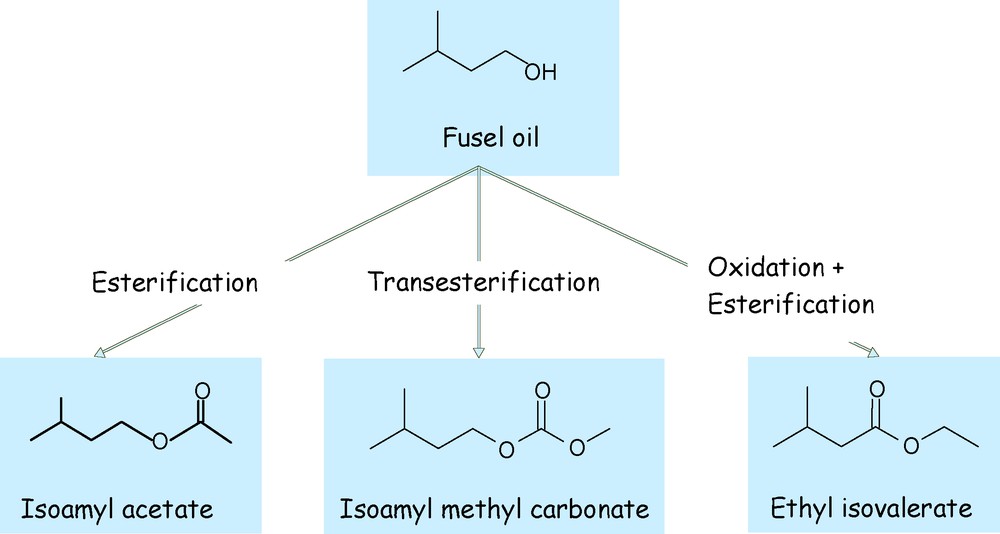
Derivatives prepared from fusel oil.
So, we started from fusel oil, which represents a cheap and renewable supply since ethanol production is a rapidly growing industry. As fusel oil can be derived from bioethanol of first generation (sugar or starch) and from second generation (lignocellulosic biomass), the volumes generated allows one to develop biosolvents for specific markets. Up to now, only a low amount of fusel oil is recovered for commercial purposes (food aroma, perfume industry).
Fusel oil corresponds to the pot residue after distillation of fermented alcohol, either dedicated for beverages or for biofuels. It consists of a mixture of alcohols, from C2 to C5, coming from the decomposition of proteins present in the plant. These short alcohols constitute between 50 and 70% of the mixture, depending on the origin (Table 2).
Composition (mass percentage of alcohol components) of several fusel oils.
| Origin & Supplier of Fusel oils | Beet from Tereos | Beet from UNGDAa | Wheat from UNGDA a |
| Ethanol (%) | 1.69 | 11.32 | 17.65 |
| Propanol (%) | 0.03 | 0.08 | 0.09 |
| Isopropanol (%) | < 0.01 | 0.05 | < 0.01 |
| Butanol (%) | 0.14 | 0.06 | 0.15 |
| Isobutanol (%) | 3.43 | 0.25 | 0.69 |
| C5 alcohols (%) b | 74.84 | 75.73 | 57.62 |
| Total (%)c | 80.13 | 87.49 | 76.20 |
a UNGDA: Union nationale des groupements de distillateurs d’alcools (France).
b C5 alcohols correspond to the mixture of isoamyl alcohol (mainly 3-methyl-1-butanol) and active amyl alcohol (2-methyl-1-butanol).
c Addition of water content allows one to reach 100%.
In this study, we proposed new synthetic processes for the production of molecules derived from isoamyl alcohol, following the principles of green chemistry. To meet industrial requirements, each method was developed for use under selective and easy-to-implement conditions.
Therefore, we determined green indicators for each new reaction from material balances. We have focused on indicators recommended by Trost and Sheldon, that is to say, the atom economy (AE), the environmental factor (EF) according to the following equations:
Mass productivity can be deduced with the following formula:
MP (%) = 100/(EF + 1) = 100/S−1 with S−1, the mass index defined by Eissen and Metzger [13].
Calculated green indicators are compared with those of usual processes carried out with isoamyl alcohol.
2 Experimental part
Several chemical derivatives have been obtained from the alcohols in fusel oil: isoamyl acetate, methyl isoamyl carbonate and ethyl isovalerate (Fig. 1).
These derived compounds have moderate volatility with vapor pressures below the values of conventional solvents (including chlorinated solvents, hydrocarbons, ketones and ethers).
Syntheses were performed with isoamyl alcohol (98%), obtained by distillation of fusel oils derived from sugar beets and wheat, in order to facilitate treatment of the reaction medium and to characterize the final products. Fusel oils, provided by UNGDA and Tereos, were collected from several French industrial distilleries. Their compositions are described in Table 2.
All other commercial reagents were used without any preliminary purification.
We determined reaction yields by gas chromatography with ChromPack 9002 apparatus equipped with a polar column CP WAX 25 m × 0.32 mm × 1.2 μm, a flame ionization detector (FID) and an on-column injector.
2.1 Materials and catalyst for syntheses
Acetic acid (99%) and acetic anhydride (99%) were purchased from Sigma Chemical company, Inc. Isoamyl acetate (> 99%) was supplied by Fisher Sciencific. The cation-exchange resin used in this study was Amberlyst® 15 supplied by Rohm & Haas. It is a macroreticular styrene divinyl benzene resin grafted with sulfonic acid groups.
The wet resin was first washed with methanol to remove any water that might be sorbed. It was then dried in a vacuum oven at 353 K for 6 h and stored in a desicator until further use. Specific surface area was 42 m2/g, determined using the BET method, with a capacity close to 4.8 meq H+/g.
2.2 Syntheses methods
2.2.1 Method 1: heterogeneous esterification of isoamyl alcohol with acetic acid
Isoamyl alcohol (454 mmol), glacial acetic acid (378 mmol) and the cation-exchange resin H+ Amberlyst® 15 (2 g) were stirred for 1 h at 120 °C. Water was continuously removed through Dean Starck. The catalyst was separated through filtration by gravity. Isoamyl acetate (47.7 g, 97%) was obtained as a colorless liquid by distillation at atmospheric pressure (T = 141–142 °C).
2.2.2 Method 2: heterogeneous esterification of isoamyl alcohol with acetic anhydride
Isoamyl alcohol (454 mmol), acetic anhydride (227 mmol) and the cation-exchange resin H+ Amberlyst® 15 (1 g) were stirred at reflux (120 °C) for 2 h to give a conversion rate of 98%. Isoamyl acetate was separated from acetic acid by distillation at atmospheric pressure.
1H NMR (300 MHz, CDCl3) : δ (ppm) = 4.05–4.09 (t, 2H, OCH2), 2.03 (s, 3H, CH3CO), 1.67 (m, 1H, CH), 1.48 (m, 2H, CH2), 0.90–0.92 (d, 6H, CH3CH).
2.2.3 Method 3: oxidation of isoamyl alcohol with hydrogen peroxide
Tungstic acid (2.2 mmol) was added, in the form of a yellow powder, to 15 mL of water and 15 mL (175 mmol) of hydrogen peroxide (35% solution in water). This mixture was heated at 55 °C until a homogeneous solution was reached. We then added 3-methyl-1-butanol (113 mmol) and 35% hydrogen peroxide (33.1 g, 340 mmol) simultaneously. The intermediate reaction was stirred at 90 °C for 24 hours. The medium was cooled at room temperature until the formation of two distinct phases. The organic phase was separated by settling. The isovaleric acid dissolved in aqueous gas phase was extracted with cyclohexane. The two organic phases were collected and dried on MgSO4. Isovaleric acid (9.2 g, 79%) as a colorless viscous liquid was obtained by distillation under reduced pressure (40 mmHg, 85 °C).
2.2.4 Method 4: esterification of isovaleric acid
Isovaleric acid (3-methylbutan-1-oic acid) (196 mmol), ethanol (392 mmol) and 9 g previously prepared exchange resin Amberlyst® 15 (cf §2.1 Materials and catalyst) were stirred at reflux for 4 h (80 °C). Ethyl isovalerate was recovered as a colorless liquid, by distillation at atmospheric pressure (T = 131–133 °C) with a 79% yield.
1H NMR (400 MHz, CDCl3): δ (ppm) = 4.12–4.14 (q, 2H, OCH2), 2.2 (d, 2H, CH2CO), 2.10–2.11 (t, 1H, CH), 1.26 (t, 3H, CH3CH2O), 0.95–0.97 (d, 6H, CH3CH).
2.2.5 Method 5: transcarbonatation of dimethylcarbonate (DMC) with isoamyl alcohol
Isoamyl alcohol (196 mmol), dimethyl carbonate (DMC) (392 mmol) and 1.7 g of K2CO3 catalyst were stirred at reflux for 2 h (90 °C). The isoamyl carbonate was recovered as a colorless liquid, by distillation at atmospheric pressure (T = 157–158 °C) with a 52% yield.
1H NMR (400 MHz, CDCl3): δ (ppm) = 4.11–4.12 (t, 2H, OCH2), 3.78 (s, 3H, OCH3), 1.69–1.71 (dd, H, CH), 1.55 (t, 2H, OCH2CH2), 0.90–0.92 (d, 6H, CH3).
For each synthesis, AE and EF have been calculated.
For the determination of EF, the following materials have been integrated for the calculation of the amount of wastes:
- • the non reactant alcohol according to the yield;
- • the excess of reactant such as acetic acid or DMC;
- • the aqueous solvent (involved in the oxidation reaction) and the organic solvents for the conventional methods;
- • the co-product such as water or methanol;
- • the catalyst (sulfuric acid, tungstic acid, K2CO3…);
- • the amount of water necessary to remove the homogeneous catalysts;
- • a quater of the macroporous resin weight, since the same lot of resin can be used four times.
These hypotheses allow one to compare the different routes to obtain the targeted product.
3 Results and discussion
3.1 Esterification
Esterification is a well-established process, commonly used in industry. Industrial processes usually involve a homogeneous catalyst (strong acid such as sulfuric acid). This approach, although tightly controlled, leads to the formation of aqueous pollutants. Therefore, various alternatives have been proposed in recent years. In particular, many ‘cleaner’ synthetic methods have been developed for the preparation of isoamyl acetate. Such methods have focused on using milder conditions at room temperature [14], involving an enzyme [15], or new catalytic systems such as ionic liquids [16], superacids [17,18], cationic resins [19] and other specific catalysts [20,21,22,23].
Acetic anhydride has also been used as an esterification agent to produce ethyl acetate and tert-butyl acetate [24,25] in a catalyst-free industrial process. This synthetic method provides a high conversion rate but leads to the formation of acetic acid, at the same time losing half the molecule of acetic anhydride, resulting in a poor AE (68%). We studied the synthesis of isoamyl acetate using a macroporous acid ion exchange resin as a heterogeneous catalyst. This method has the advantages that the reaction medium is easier to handle and the catalyst can be reused without generating acid effluents.
Two new methods of isoamyl acetate synthesis were studied in the presence of an acid ion exchange resin:
- • esterification of isoamyl alcohol with acetic acid;
- • esterification of alcohol with limiting acetic anhydride.
Esterification of acetic acid was thus performed with a catalyst loading of 5% w/w (i.e. weight of dried Amberlyst® 15/weight of alcohol), corresponding to 2% of H+ equivalents. We then determined the effects of variables such as reaction temperature, molar ratio of reactants and reaction time (Table 3). Moreover, as the acid-catalysed esterification reaction is an equilibrium-limited chemical reaction, the equilibrium shift was performed by removing water during its formation (reactions no 7, 8 and 9).
Esterification of acetic acid under heterogeneous catalysis.
| No | Reaction temperature | Molar ratio alcohol:acid | Reaction time (h) | Yield (%) |
| 1 | 70 | 1:1 | 1 | 9 |
| 2 | 70 | 1:1 | 4 | 65 |
| 3 | 100 | 1:1 | 1 | 22 |
| 4 | 100 | 1:1 | 4 | 66 |
| 5 | 120 | 1:1 | 1 | 67 |
| 6 | 120 | 1:1 | 4 | 67 |
| 7a | 120 | 1:1 | 1 | 81 |
| 8a | 120 | 1.2:1 | 1 | 97 |
| 9a | 120 | 2:1 | 1 | 97 |
a equilibrium shifted by Dean-Stark apparatus.
These results show that a high temperature facilitates the crossing of the activation energy barrier to reach the reaction equilibrium faster.
The removal of water and the addition of a slight excess of isoamyl alcohol contribute to the equilibrium shift of the reaction, thus obtaining a better performance level (experiment 8). The unconverted isoamyl alcohol can be recycled to improve both the environmental and economic aspects of this method.
Amberlyst® 15 was reused up to four times (each time for 4 h run), without loss of its catalytic activity. A slight reduction in yield (8%) was only observed at the second reaction.
Synthesis of isoamyl acetate from acetic anhydride without the use of a catalyst leads to a high conversion rate for isoamyl alcohol, but generates an equimolar amount of acetic acid, making purification from the medium difficult (Table 4, experiment 10). Moreover, any trace of acetic acid confers a stinging odor to isoamyl acetate. To lessen these effects, the two reactions were combined as shown in Fig. 2: esterification with acetic anhydride and esterification with acetic acid in the presence of a heterogeneous catalyst.
Esterification of acetic anhydride acid with isoamyl alcohol.
| No | Reaction | Molar ratio alcohol:anhydride | Reaction time (h) | Catalyst amounta (%) | Yield (%) |
| 10 | 100 | 1:1.1 | 1 | – | 99.8 |
| 11 | 120 | 1:0.5 | 2 | 2.5 | 98 |
a Dried Amberlyst® 15, mass percentage relative to alcohol.
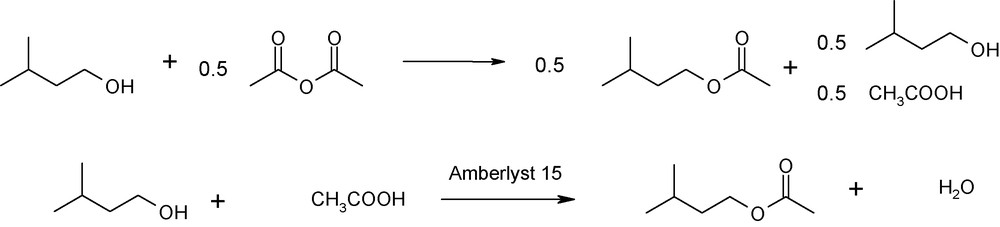
Equation for esterification, with acetic anhydride as the limiting reactant.
Both reactions can be implemented in an one-pot process, as shown in Fig. 3.
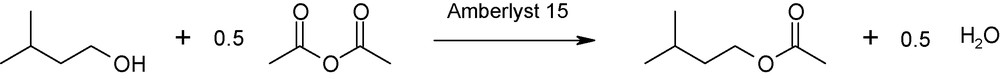
Esterification reaction with optimized atom economy.
These conditions were experimented with assay 11 in Table 4.
Our results show a quantifiable yield after two hours of reaction. This is associated with reduced amounts of reactant and of waste produced, both aspects being beneficial for industry. It thus represents a new approach for the esterification reaction.
We compared the sustainability of the studied reactions with that of the industrial process currently in general use to convert primary short alcohols (Table 5).
Comparison of the green indicators for esterification reactions.
| Reagent | Catalyst | Atom Economy (%) | Environmental factor E | Mass productivity (%) | Waste | ||
| Industrial conditionsa | Homogeneous catalyst | CH3COOH | H2SO4 | 88 | 2.5 | 29 | Acidic waste (H2SO4 + CH3COOH) |
| Method 1 Assay 8 Table 3 | Heterogeneous catalyst | CH3COOH | Cation exchange resin | 88 | 0.3 | 78 | None |
| Assay 10 Table 4 | No catalyst | Acetic anhydride | No catalyst | 68 | 1.1 | 48 | Acidic waste (CH3COOH) |
| Method 2 Assay 11 Table 4 | Heterogeneous catalyst | Acetic anhydride | Cation exchange resin | 94 | 0.07 | 93 | None |
a Experimental conditions: 80 °C, 2h, 2% eq.H+ catalyst, molar ratio alcohol:acetic acid = 1:1, Yield: 69%.
In the present case, the use of ion exchange resin provides the means for a cleaner and easier synthetic process, with high AE. The conditions used for experiment 11 generated a reduced amount of waste (lower environmental impact factor) and avoided the need for treatment of aqueous effluents. Removal of this treatment step contributes to the gained energy.
Mass productivity serves as an essential factor to be taken into account for evaluation of the relative benefits of a process. The only waste produced in experiment 11 is 70 g of water for the preparation of 1000 g of isoamyl acetate.
3.2 Synthesis of ethyl isovalerate
This synthesis involves two steps: the first step involves the oxidation of isoamyl alcohol to form valeric acid and the second step is an esterification reaction to form ethyl isovalerate (Fig. 4).

Preparation of ethyl isovalerate.
3.2.1 Isoamyl alcohol oxidation
Traditionally, the oxidation of primary alcohol to form carboxylic acid involves the use of stoichiometric inorganic oxidants, such as a chromium (VI)-based reactant [26], manganese or ruthenium salts [27]. Such catalysts are highly toxic for the environment and incompatible with cosmetic applications. Therefore, with legislation on heavy metals becoming more and more stringent, these catalysts are no longer used. Oxidation methods also involve the use of toxic reactants, for example periodinane and oxalyl chloride [28].
To meet economic and environmental demands, other more recent methods use clean oxidants such as dioxygen, in the presence of TEMPO (2,2’,6,6-tetramethyl piperidine-N-oxyl) or PIPO (polymer-immobilized piperidinyl oxyl).
The use of hydrogen peroxide is of particular interest because water is the only theoretical co-product. Moreover, aqueous H2O2 can be considered as a clean oxidant if no organic solvent or any other toxic compound is used [29]. Furthermore, the use, storage and transportation of low concentrated H2O2 are not problematic in terms of safety. The most effective catalysts for oxidation with H2O2 tend to be early transition metals ions with a d0 configuration, such as MoVI, WVI, ReVII for homogeneous catalysts and TiIV, a heterogeneous catalyst. Such elements generally display low catalytic activity, preventing H2O2 decomposition.
However, most of these methods are not directly aimed at the synthesis of the carboxylic acid but rather are selective for the synthesis of the aldehyde. Noyori et al. have reported the efficiency of H2O2 coupled with a tungstate complex and an acidic phase transfer catalyst, for the oxidation of primary alcohol [30]. Therefore, we tested the catalytic system H2WO4/H2O2 in the oxidation of isoamyl alcohol (Fig. 5).
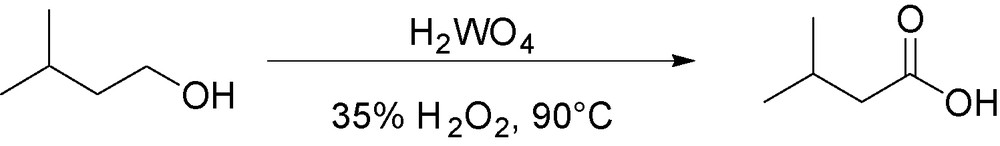
Selected conditions for isovaleric acid synthesis.
Hydrogen peroxide is used as a clean and cheap oxidative agent in the oxidation of propylene by BASF and Dow Chemical and for the synthesis of caprolactone by Sumitomo Chemicals Co. Diluted in water to below 60%, it maintains its efficiency over several months and can be stored without risk. Moreover, tungsten occurs naturally as tungstate (WO42−) and its level of toxicity is rather lower than other heavy metals (International Tungsten Industry association) [31].
Various catalyst amounts and reaction times were tested to determine the appropriate conditions.
A phase transfer catalyst (PTC) was only added for assay 7 (Table 6).
Oxidation with oxygenated water: effect of catalyst amount and reaction time on isovaleric acid production.
| H2WO4 (% mol./ROH) | Reaction time (h) at 90 °C | Yield (%) | |
| 1 | 1 | 7 | 11 |
| 2 | 2 | 7 | 40 |
| 3 | 5 | 7 | 45 |
| 4 | 10 | 7 | 45 |
| 5 | 5 | 24 | 79 |
| 6 | 5 | 30 | 80 |
| 7 | 2 + PTC a | 7 | 82 |
a PTC = Aliquat 336 (2% mol. /ROH).
Our results show that at least 5% mol/ROH of catalyst is needed for oxidation of isoamyl alcohol, giving a 79% yield. Overall selectivity for the production of isovaleric acid was observed after 24 hours. This process is simple to operate, with hydrogen peroxide acting both as a reactant and a reaction solvent, which solubilizes catalyst. The oxidizing complex in this reaction is H2[WO(O2)2(OH)2], which is water-soluble and has a pKa of around 0.1. The inclusion of 2% methyltrioctyl ammonium salt (Aliquat® 336) as a phase transfer catalyst (PTC) reduced reaction time from 24 h to 7 h, with a 82% yield, reflecting a better conversion rate. However, the addition of PTC also leads to a more difficult separation of the product.
This method was compared to conventional oxidation reactions in Table 7.
Comparison of green indicators for oxidation reactions.
| Method (reference) | Substrate | Oxidizing agent | Catalyst | Solvent | Atom Economy (%) | E-factor | Hazards |
| [32] | Isoamyl Alcohol | NaOCl | CH3CN /CH3COOH CH2Cl2 | 62.9 | 182 | Generation of chlorinated derivatives | |
| [33] | n-amyl Alcohol | NaOCl NaOCl2 | TEMPO | CH3CN | 57.1 | 58 | Formation of salts and chlorinated derivates |
| [34] | 1-octanol | NaOCl | PIPO | hexane | 70.4 | 91 | Generation of chlorinated derivatives |
| Method 3 | Isoamyl Alcool | H2O2 (35%) | H2WO4 | no | 83.6 | 6.7 | Aqueous solution of H2WO4 |
| Essay 5 | |||||||
| Table 6 |
The use of the selected catalytic system H2WO4/H2O2 is an efficient method in terms of AE and leads only to aqueous effluents.
Despite using a non-toxic catalyst, the usual oxidation reaction with TEMPO does not respect the whole green principles (waste production, solvent use) compared to the studied path.
3.2.2 Esterification of isovaleric acid
The synthesis of ethyl isovalerate from isovaleric acid involves an esterification reaction with ethanol. This step was carried out using similar conditions as used for isoamyl alcohol esterification. The reaction was performed with ion exchange resin catalysis in refluxing ethanol, obtained directly from the oxidation medium containing isovaleric acid and less than 20% of non-reacted isoamyl alcohol. No intermediate step of purification was therefore needed, keeping the process simple to operate. We studied the effects of changes in ethanol:acid ratio and amount of resin on the conversion rate. Results are presented in Table 8.
Conversion of isovaleric acid according to stoichiometry.
| Assay | Resin weight (%mass/ethanol) | Molar ratio (ethanol:isovaleric acid) | Conversion vs. isovaleric acid after 4h (%) | Conversion vs. isovaleric acid after 7h (%) |
| 1 | 5 | 1:1 | 50.4 | 53.6 |
| 2 | 5 | 1:2 | 30.6 | 35.7 |
| 3 | 5 | 3:1 | 24.9 | 28.2 |
| 4 | 5 | 5:1 | 16.2 | 26.0 |
| 5 | 5 | 10:1 | 6.5 | 12.3 |
| 6 | 50 | 1:1 | 77.3 | – |
| 7 | 50 | 1.5:1 | 80.7 | – |
| 8 | 50 | 2:1 | 85.6 | – |
| 9 | 50 | 3:1 | 74.8 | 83.6 |
| 10 | 50 | 5:1 | 56.1 | – |
| 11 | 50 | 10:1 | 41.7 | – |
| 12 | 75 | 3:1 | – | 86.8 |
We observed a decrease in conversion rate with increasing alcohol:acid molar ratio, which can be explained by an increasing dilution factor of the resin in the reaction medium. A high amount of resin led to a higher conversion of isovaleric acid (85.6%) for a 2:1 alcohol:acid molar ratio. This excess of alcohol shifted the reaction equilibrium, because the water could not be distilled during the reaction due to a water:ethanol azeotrop (with a 95.6:4.4 composition at 78.2 °C). The reaction was selective for the production of ethyl isovalerate, in all conditions tested. No trace of isoamyl isovalerate was detected by gas chromatography.
Ethyl isovalerate can be isolated by distillation (boiling point = 132–134 °C). This was the only purification step required after successive oxidation and esterification, leading to a global yield of 67% after a total reaction time of 11 hours.
3.3 Isoamyl alcohol carboxymethylation
The selective synthesis of unsymmetrical carbonate is difficult due to the formation of the more stable symmetrical carbonate (diisoamyl carbonate).
Many methods for the synthesis of alkyl carbonates from alcohols have thus been exploited. Reactants such as urea or its carbamate derivatives [35], carbon monoxide [36] or supercritical CO2 [37,38], lead to low yields for the preparation of mixed carbonates. The selectivity for mixed carbonates can be improved using more nucleophilic and unsymmetrical reagents such as halogenoalcanes [39], acid halides [40] or haloformates [41]. These syntheses involve polar solvents such as acetonitrile or dimethylformamide.
However, these unsymmetrical carbonates are still prepared on an industrial scale from highly toxic starting materials (such as phosgene COCl2) via methyl chloroformiate. New pathways involve carbonated building blocks such as DMC, which has been shown to be an efficient methoxycarbonylating agent and can be formed by a clean synthetic process [42,43].
However, the transesterification of aliphatic alcohols with DMC is still carried out with toxic catalysts such as (n-Bu2SnO)6 [44] or requires a phase transfer catalyst with high temperature and pressure conditions [45].
Recent work by Tundo et al. [46] showed the efficiency of such a reaction using K2CO3: a high temperature (200 °C) and a considerable excess of DMC were needed to obtain a good yield and high selectivity. These conditions require a batch autoclave, which is not suitable for large-scale industrial production.
We adapted this process for the methoxycarbonylation (also called transcarbonatation) of isoamyl methyl carbonate with DMC, through a simpler process and under milder conditions. This reaction is based on the transesterification mechanism. DMC, doted of high polarity, serves as both a solvent and a reactant.
The mixed carbonate was formed through the BAC2 mechanism (step 1, Fig. 6). The distillation of methanol to shift the equilibrium is only recommended to produce diisoamyl carbonate (step 2).
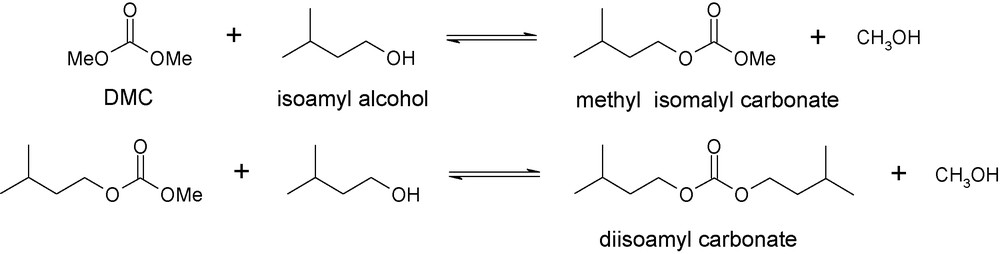
Transesterification of isoamyl alcohol by DMC.
3.3.1 Influence of catalyst
Basic catalysts are effective in the preparation of a mixed carbonate from DMC and an alcohol. However, the use of metal catalysts can generate toxic traces of heavy metals in the final product, requiring costly purification to meet legislative requirements.
Therefore, we tested non-metallic catalysts: two inorganic alkali compounds such as sodium hydroxide NaOH or potassium carbonate K2CO3, and an organic compound such as sodium methylate MeONa, which behaves as a homogeneous catalyst.
The results presented in Table 9 show high conversion and selectivity rates for the three catalysts. Moreover, these conditions can minimize the formation of the symmetrical carbonate. The kinetics of the reaction with potassium carbonate was twice as slow as those with the other catalysts; however, the use of potassium carbonate also resulted in a higher selectivity for mixed carbonate production.
Effect of catalysis on reaction performance.
| Assay | Catalyst (1 %) w/w vs alcohol) | Type of catalyst | Reaction time (h) | Conversion (%) | Selectivity (%) | Yield (%) |
| 1 | NaOH | Heterogeneous | 1 | 84 | 65 | 55 |
| 2 | MeONa | Homogeneous | 1 | 90 | 60 | 54 |
| 3 | K2CO3 | Heterogeneous | 1 | 54 | 74 | 40 |
| 4 | K2CO3 | Heterogeneous | 2 | 82 | 71 | 58 |
Despite its effectiveness, the use of sodium methylate is not compatible with the principles of green chemistry. Its miscibility in the reaction medium complicates its disposal since a washing step with water is needed, involving the production of large quantities of alkaline waste products. Additionally, it is not reusable and its low stability during storage (hydrolysis in methanol) makes its application in industry difficult.
NaOH and K2CO3 are easily separated from the final medium because they are not soluble. Moreover, they are relatively inexpensive and have only very low levels of toxicity. However, potassium carbonate is a better desiccant than NaOH, allowing both the catalysis of the reaction and the capture of water that may be present in the reaction medium if fusel oil is directly employed as reactant (Fig. 7).
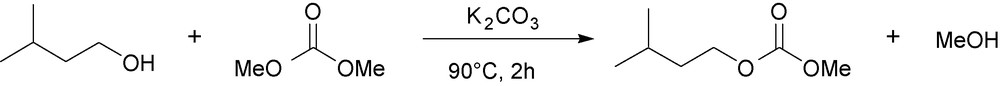
Selected conditions for the carbonatation of isoamyl alcohol.
3.3.2 Influence of molar DMC:alcohol
We tested several molar ratios of DMC:alcohol for increased selectivity of the reaction. Our results (Table 10) showed that an excess of DMC favored the reaction selectivity. The DMC:alcohol ratio was set at 5 to limit dilution of the medium and to maintain a high conversion rate.
Effect of the DMC: alcohol molar ratio on the reaction selectivity.
| Ratio DMC: alcohol | 2:1 | 3:1 | 5:1 |
| Conversion (%) | 82 | 79 | 75 |
| Selectivity (%) | 71 | 83 | 95 |
| Yield (%) | 58 | 66 | 74 |
The use of non-distilled fusel oil containing 7% water affects the catalytic activity of potassium carbonate, reducing the reaction rate, so that 10 hours are needed to obtain the maximal yield (74%), rather than the 2 hours indicated in Table 10.
The catalyst was filtered and the medium distilled to separate the different compounds.
Table 11 shows the composition of the collected fractions.
Fractions collected by distillation after methoxycarbonylation of isoamyl alcohol.
| Fraction | Temperature (°C) | Composition | Fate |
| 1 | 63.5 | Methanol:DMC (70:30) | Difficult purification → waste |
| 2 | 90 | DMC (99%) | Recyclable |
| 3 | 132–134 | Isoamyl alcohol (98%) | Recyclable |
| 4 | 157–158 | Methyl isoamyl carbonate (98%) |
An energy saving synthetic process was thus developed with a methoxycarbonylating agent (DMC), which is biodegradable and non toxic [45]. This process involves heterogeneous conditions and uses a five-fold excess of refluxing DMC at 90 °C for a reaction time of one hour.
A large excess of DMC prevents the formation of symmetrical carbonate and leads to the production of isoamyl methyl carbonate with high selectivity (95%). Distillation is the only purification step required to obtain methyl isoamyl carbonate. Reaction conditions are compatible with large-scale transfer by an economically viable process.
We compared characteristics of green chemistry between the various methods of isoamyl alcohol methoxycarbonylation (with DMC or phosgene) (Table 12).
Green indicators of carboxymethylation methods.
| No | Reagent | Catalyst | Solvent | Atom economy (%) | E-factor | Mass productivity (%) | Hazards |
| [47] | Phosgene | Pyridine | Hexane | 74.7 | 5.4 | 16 | Toxic reactant |
| Assay 2 | DMC | MeONa | – | 82 | 3.4 | 23 | Aqueous basic waste |
| Table 9 | |||||||
| Method 5 Assay 4 Table 9 | DMC | K2CO3 | – | 82 | 1.3 | 42 | Low impact |
Reactions involving DMC have reduced effects on the environment. Moreover, the protocol for heterogeneous catalysis does not involve toxic substances and avoids the production of waste products.
Method 5 is thus a safe alternative for the selective carboxymethylation of alcohol. This process seems to offer the best compromise taking into account economical aspects, performance and environmental concerns.
3.4 Comparison of volatility characteristics of synthesized biosolvent
Flash point and boiling point are two important characteristics affecting the drying time of nail polish formulations. Several formulations have been prepared with the synthetized solvents and the conventional solvents. They consist of the following ingredients: 10.5% of nitrocellulose (polish agent), 5% of acetyl tributyl citrate (plastizicer), 7.5% of polyester resin and 77% of solvent.
Table 13 shows the drying time of these nail polish formulations. We compared the data of the new formulations with those of the standard formulation prepared with a blend of ethyl acetate and butyl acetate.
Volatility characteristics for solvents and mixture of solvents used for the nail polish formulations.
| Solvents | Formulation flash point (°C) | Solvent boiling point (°C) | Time until dry to touch (min) | Total drying time (min) |
| Butyl acetate/ethyl acetate (40/ 60 w/w) | 3 | 125/77 | 3′ | 3′30 |
| Isoamyl acetate | 25 | 125 | 12′ | 13′30 |
| Ethyl isovalerate | 26.7 | 135.1 | 13′ | 15′ |
| Isoamyl and methyl carbonate | 55.5 | 157 | 16′ | 21′ |
The drying times of the biosolvent formulations were longer than those observed for the standard formulation (3′). However, they are still acceptable for the intended use when a cosolvent is added in the formulation [5].
We calculated the resulting VOC emissions of the biosolvent with the faster drying time (isoamyl acetate).
3.5 Evaluation of VOC emissions during the production of a nail polish
The amount of VOC released into the atmosphere during the production of solvent-based products is controlled by environmental regulations. Solvent evaporation is seen at each stage of the commercial production of coatings (for example, nail polish, paint or ink). Photochemical interactions between VOC and nitrogen oxides (NOx) then generate ground-level ozone, responsible for bad air quality. To help industry reduce their VOC emissions, the U.S. Environmental Protection Agency has published guidelines describing techniques for estimating the emissions produced during the manufacture of paints, inks and other coatings [48]. These methods have been applied to processes currently in use for the manufacture of coatings, to estimate annual VOC emissions due to solvent use. The generation of emissions has been assessed for surface evaporation during dispersion of the ingredients in an open tank according to the Eq. (1) and Table 14. It is one of the step which releases the highest proportion of emissions into the atmosphere.
| (1) |
List of parameters used in Eq. (1).
| Symbol | Variable | Production parameters |
| Ex | Emissions of VOC species | kg/yr |
| Kx | Gas-phase mass transfer coefficient for VOC species | m/sec |
| Mx | Molecular weight of VOC species | g/mol |
| A | Surface area of tank opening | 10.2 m2 |
| Px | Partial vapor pressure of chemical x at temperature T | kPa |
| H | Batch time necessary to perform the blend | 3 hr |
| R | Universal gas constant at a pressure of 1atm | 8.31 J/mol/°C |
| T | Ambient Temperature | 20 °C |
| B | Number of batches per year | 300/yr |
The formulation of a conventional nail polish formulation, with a volatile fraction containing three petrochemical solvents (ethyl acetate, butyl acetate and isopropanol) is then compared with the production of a nail polish formulation in the same conditions using natural-based solvents (Table 15).
Estimated VOC emissions during the dispersion step of formulations (6000 tons) for a full year.
| Formulation | VOC emission (kg/year) | Emissions decrease compared to standard (%) |
| Standard nail polisha | 1262.8 | 0 |
| Isoamyl acetate | 163 | −87 |
| Fusel acetate | 406.9 | −68 |
a Volatile fraction composition: ethyl acetate 57.5%; butyl acetate 23.8%; isopropanol 18.7%.
The results show that the use of alternative solvents from fusel oil contributes to reduce VOC emissions of at least 68%. Such solvents thus provide a solution for reducing the effects on the environment.
The modeling equations can be used to select an appropriate solvent and to estimate the corresponding emissions, before more complex analysis is required.
4 Conclusion
This study has demonstrated that a co-product of the bioethanol industry can be used as a synthon to produce safer molecules for the replacement of solvents currently in use.
Conventional methods such as esterification, oxidation or transcarbonatation have been modified into ecofriendly processes. External solvent-free reactions, heterogeneous catalysis, mild conditions and straightforward purification are the main parameters which lead to improved green metrics.
We have shown that green units represent an useful concept to select the valuable molecules as efficient competitors towards the petrochemical molecules.
Moreover, VOC emission models provide key tools to check the environmental benefits for the resulting biobased formulations.
Eventually, the molecules and the associate processes which match the targeted criteria for the development of new biosolvents, deserve a further study for a scale transfer of their production to industry.
Acknowledgement
Durlin company is gratefully acknowledged for their financial support and for their cooperation.