

1 Introduction
Calculation of magnetic coupling constants in molecules and materials from a purely theoretical basis is a common practice among computational chemists and physicists. There exist a few phenomenological spin Hamiltonians (Ising, Heisenberg, etc.) that relate the energies of different spin states with the J parameter, which quantifies the strength of the magnetic interaction. Several methodologies based on quantum mechanics have been applied to compute J, all with their strengths and weaknesses. For small to medium sized systems (either molecules or model fragments of crystals), highly correlated ab initio (wavefunction based) methods can be applied, notably the difference dedicated configuration interaction (DDCI) method [1,2], developed to accurately calculate energy differences. In general, and especially for large systems, a balance between computational time and accuracy is a must.
In this article, we apply the density functional theory (DFT) to the calculation of the J parameter in systems containing 90 to 130 atoms. Apart from one study in which ab initio correlated methods were applied [3], DFT based approaches are commonly used to calculate J in extended metal atom chains (EMACs) [4], our target systems in the present work. Such molecules contain a set of metal atoms disposed in a linear arrangement thanks to the coordination to polydentate organic ligands of the polypyridylamide family [5], of general formula [C5H4N-(N-C5H3N)n-N-C5H4N](n+1)− and number of binding sites p = 2n + 3, cylindrically wrapped around the chain. The number of binding sites of the purposely-designed organic ligands determines the nuclearity of the metal chain. The homonuclear trimetallic chains, M3(dpa)4Cl2, are the most common examples of EMACs [6–17]. A recurring challenge related to metal string complexes is the synthesis of increasingly longer chains [18–26]. Peng's group achieved an 11-nickel molecular string, the longest metal chain known in 2011 [27,28].
Very recently, we calculated the magnetic coupling constant for Ni3(dpa)4Cl2 and some hypothetical derivatives [29] using the method reported by Labèguerie et al. [30]. The previously proposed decomposition of the J parameter in this compound, J = Jσ + Jδ, was carried out, where each component arises from a different coupling pathway within the molecule. The motivation of the present work arose a few years ago, when Rohmer and Bénard obtained an unexpected DFT underestimation of the J parameter for Ni3(dpa)4Cl2 [4a], a well-known system whose magnetic properties have been characterized [26,31,32]. The smaller value computed contrasts with the typical overestimation of J for the other members of the EMAC family, notably [Cu3(dpa)4Cl2]+, and most magnetic systems theoretically studied at the same level of calculation. We herein present calculations on [Cu3(dpa)4Cl2]+, Ni3(dpa)4Cl2 and Ni5(tpda)4Cl2 (dpa: dipyridylamido; tpda: tripyridyldiamido), shown in Fig. 1, and discuss why DFT/B3LYP results on Ni derivatives tend to underestimate J in contrast to other compounds of this class.
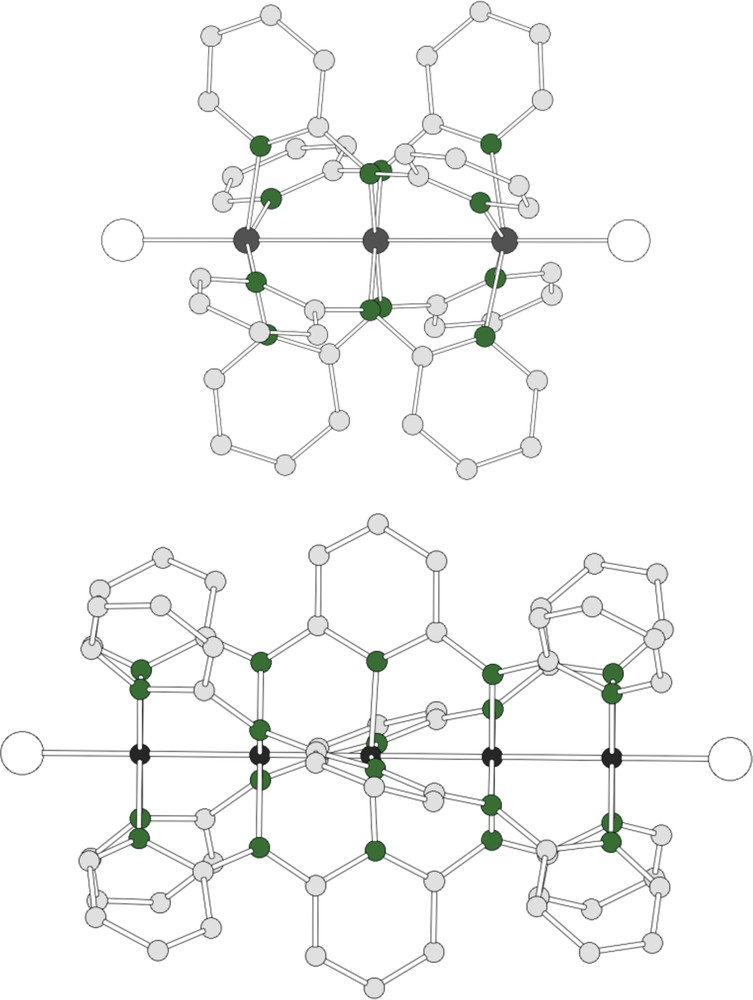
Structure of M3(dpa)4Cl2 (M = Cu, Ni) (top) and Ni5(tpda)4Cl2. The central backbone contains the metal atoms, in grey. Green spheres represent nitrogens. Hydrogen atoms have been omitted for clarity.
2 Computational details
The Gaussian03 [33] suite of programs was used to calculate all the spin state energies using the broken symmetry technique of Noodleman et al. [34,35]. We used Becke's gradient corrected three-parameter B3LYP functional [36], of the form
| (1) |
There has been a great deal of controversy [42–44], in the literature, regarding the use of spin projection with density functionals since the self-interaction error (SIE) [45] tends to mimic static correlation, therefore, making the Kohn-Sham broken symmetry determinant artificially represent more than just half of an open shell singlet. While most hybrid density functionals should carry less SIE with respect to their “pure” counterparts most additions of exact exchange in hybrids are usually modest and do not generally exclude enough SIE to guarantee a sufficient accuracy with spin projection [46]. It should be noted that the formalism devised by Labèguerie et al. [30] does in fact presuppose spin projection in all the broken symmetry solutions.
3 Results
The systems we deal with present two magnetic centres located at both ends of the chain. The central metal atoms (one for M3 chains, three for M5 chains) are diamagnetic due to the ligand field of their neighbouring atoms. In the case of [Cu3(dpa)4Cl2]+, the terminal d9 copper atoms posses a local S = 1/2, whereas for Ni3(dpa)4Cl2 and Ni5(tpda)4Cl2, terminal d8 nickel centres have local S = 1. The coupling mechanism in copper complexes is rather familiar, whereas the interaction of two 3d8 Ni(II) centres may be viewed as an interference phenomenon between two half-filled fragment molecular orbitals of eg* symmetry that are composed mainly of the 3dz2 (σ) and 3dx2–y2 (δ) atomic orbitals, yielding linear combinations of gerade and ungerade symmetries. This magnetic interaction can be phenomenologically described by a general form of the magnetic spin Hamiltonian [47–49] that has been successfully applied to single determinant methods such as DFT, more specifically to S = 1 magnetic dimers [29,30].
This Hamiltonian takes the form:
| (2) |
| (3) |
| (4) |
| (5) |
In this formalism, tσ stands for the hopping integral related to the energy splitting of the in- and out-of-phase σ orbital pair as described, for instance, in the Hay-Thibeault-Hoffmann [50] formalism. Analogously, tδ is the hopping integral for the δ symmetry pair. U is the promotion energy to attain the ionic configuration with respect to the neutral form, and Kσδ is the direct exchange integral between the σ and δ-type orbitals on the same site. λij is the biquadratic exchange constant and Bij the three-body interaction constant. In the case of simple bilinear exchange, only (Bij = λij = 0) Eq. (2) reduces to the more familiar Heisenberg-Dirac-van Vleck Hamiltonian [43] in which Jij,ef > 0 for ferromagnetic compounds and Jij,ef < 0 for anti-ferromagnetic interactions.
For the three complexes Ni5(tpda)4Cl2, Ni3(dpa)4Cl2 and [Cu3(dpa)4Cl2]+, the nature of the frontier orbitals involved in the magnetic coupling is depicted in Fig. 2. The magnetic centres are the outermost metal atoms in all the cases. The first two constitute a four-electron/four-orbital problem whereas the latter is a simpler two-electron/two orbital-problem. The orbitals classified as δ in Fig. 2 are responsible for the Jδ magnetic coupling, and those classified as σ participate in Jσ. The two pathways are theoretically well known from previous studies [4b,29].
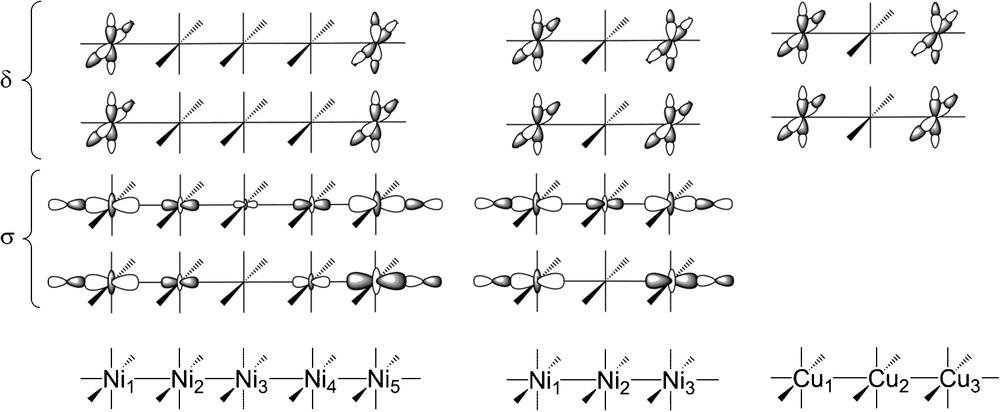
Sketch of the magnetic orbitals for the three complexes Ni5(tpda)4Cl2, Ni3(dpa)4Cl2 and [Cu3(dpa)4Cl2]+. The orbital overlap is much smaller in the σ channel than in the δ one.
A second-order perturbative treatment of the energy allows extracting electronic structure parameters related to the magnetic properties of molecules. Let us take the S = 1 systems (Ni3 and Ni5) and vary the occupation of the magnetic orbitals of σ and δ nature with ↑ and ↓ spins. We obtain the set of open-shell configurations listed in Table 1, whose single determinant representations can be classified as follows: one Ms = 2 solution (Q) two Ms = 1 solutions (T1 and T2) and two Ms = 0 solutions (S1 and S2). The perturbative treatment of their energies leads to Eqs. (6)–(10), which contain the electron structure parameters t, U and K. These values can be extracted if the energies of the states listed are known. In the present article, energies are obtained from DFT calculations. Our main goal is to calculate separately the two contributions to the total J parameter, which for an antiferromagnetic system with two S = 1 centres are, at the second order of perturbation:
Total Ms, spin orderinga and associated second-order perturbative energy expressions for the DFT solutions calculated. The energy of the quintet state is taken as zero.
| Ni1 | Ni3/Ni5 | ||||||
| Ms | Electronic configuration | σ | δ | σ | δ | E(2) | |
| 0 | “Singlet” 1: S1 | ↑ | ↑ | ↓ | ↓ | −2tσ2/U − 2tδ2/U | (6) |
| 0 | “Singlet” 2: S2 | ↓ | ↑ | ↑ | ↓ | −2tσ2/U − 2tδ2/U + 2Kσδ | (7) |
| 1 | “Triplet” 1: T1 | ↑ | ↓ | ↑ | ↑ | −2tδ2/U + Kσδ | (8) |
| 1 | “Triplet” 2: T2 | ↓ | ↑ | ↑ | ↑ | −2tσ2/U + Kσδ | (9) |
| 2 | Quintet: Q | ↑ | ↑ | ↑ | ↑ | 0 | (10) |
a Ni1 and Ni3/Ni5 are the terminal atoms in nickel chains.
where AF and F stand for antiferromagnetic and ferromagnetic, respectively. This is the common way to obtain the total J from broken symmetry DFT calculations. To decompose the final expression J = Jσ + Jδ into the two components, we can make use of Eqs. (6)–(8) from Table 1:
| (11) |
| (12) |
| (13) |
The values extracted from Eqs. (12) and (13) provide the individual antiferromagnetic contributions to the total Jeff, namely Jδ and Jσ, respectively. The value of U could not be presently computed using the closed-shell solution and its second-order perturbative energy expression since the occupations of the magnetic orbitals are different from those shown in Table 1. In the present nickel derivatives, the stable closed-shell configuration corresponds to (σnb)2(σ*)2, with empty magnetic δ orbitals. Thus, an undetermined ΔE(δ − σ) parameter appears, whose extraction is not straightforward by single determinant techniques and requires the use of effective Hamiltonian approaches and ab initio calculations. Consequently, tσ and tδ cannot either be extracted from the present analysis since they require the knowledge of U. Summarizing, from Table 1, only three equations are needed to obtain the parameters we are looking for in each system, the other two being redundant1.
3.1 [Cu3(dpa)4Cl2]+
We will now turn to the three complexes to see how the B3LYP functional fares in describing all the magnetic coupling parameters heretofore described. The analysis of the copper complex is straightforward since it consists of two S = ½ magnetic centres and the coupling pathway involving the δ orbitals depicted in Fig. 2 and J can be simply derived from the energies of the ferromagnetic determinant (↑···↑) and the antiferromagnetic (↑···↓) broken symmetry solution. The experimental value of J = –34 cm−1 in [Cu3(dpa)4Cl2]+, whereas the B3LYP value is approximately double (–63.9 cm−1). This is the typical trend observed in DFT/B3LYP calculations of J applying spin projection. In the copper system, the values of J vary dramatically (by over 200 cm−1) across the range 0–30% HFX in the expected way (see Fig. 3a) that is, the coupling constant decreases when more HFX is added to the functional. The chart shows that less %HFX favour the stabilisation of the singlet broken symmetry solution. The variation of J features a smooth exponential decrease reaching a value close (–39.0 cm−1) to the experimental one (–34 cm−1) at 25% HFX. The previously reported overestimation of J by DFT/B3LYP (–64 cm−1) is herein reproduced [4b].
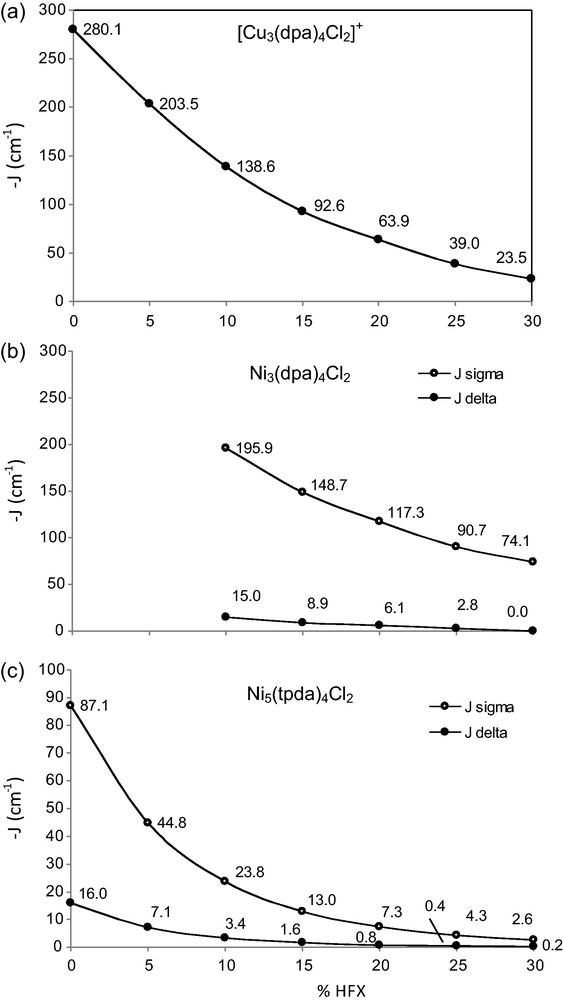
Dependency of –J (cm−1) versus the percentage of exact exchange (%HFX) for (a) [Cu3(dpa)4Cl2]+, (b) Ni3(dpa)4Cl2 and (c) Ni5(tpda)4Cl2. The vertical scale in (c) is different.
3.2 [Ni3(dpa)4Cl2] and [Ni5(tpda)4Cl2]
Fig. 3b-c contains the values of Jσ and Jδ computed for the nickel derivatives with different %HFX in the density functional. The x-axis origin corresponds to BLYP. In the case of the Ni3(dpa)4Cl2 complex, it has been shown before that the σ pathway largely dominates whereas the contribution from the δ path is tiny in comparison [29]. Supposedly, the latter is overestimated by generalization2 of the tricopper case and many other DFT studies featuring the same type of covalent metal-ligand interaction. However, it is worth noting that the sum of both contributions at 20% HFX (B3LYP) is Jcalc = –123.4 cm−1, clearly below the experimental value (–218 cm−1). Given the fact that the variation in Jσ is much larger than for Jδ, it is interesting to pinpoint the origin of such apparently incoherent behaviour of the computed J values for different compounds. If we analyze the variation of Jσ and Jδ in Ni3(dpa)4Cl2 upon changes in the amount of HFX (Fig. 3b-c), we see that the rates at which Jσ and Jδ grow are very different. The absolute change of both contributions is –6 to–15 cm−1 and –117 to–196 cm−1 for the δ and σ pathways, respectively. At smaller amounts than 20% of HFX, the dominating σ pathway resembles the experimental value. It may be deduced that the B3LYP functional overestimates the tiny Jδ but underestimates the largely dominant Jσ, overall underestimating J.
The discrepancy between the calculated and experimental J found in the Ni3(dpa)4Cl2 complex with B3LYP is also observed in Ni5(tpda)4Cl2. The pentanickel compound has an experimental J = –33.5 cm−1, well beyond the spin projected value of –8.1 cm−1 obtained with the B3LYP functional, with Jσ = –7.3 cm−1 and Jδ = –0.8 cm−1, in line with the trend observed for Ni3(dpa)4Cl2. The amount of HFX has to be reduced to ∼10% also in the computation of the Ni5 coupling constant to reproduce the experimental data (see Table 2 and Fig. 3). This establishes a rational trend in the calculation of J values in compounds featuring the Jσ – and Jδ – coupling parameters, such as the nickel derivatives herein discussed. The spin densities as a function of the exact exchange introduced vary as expected, that is, more delocalisation from metal to ligands in BLYP than at 30% HFX, following the variation of J.
Electronic structure parameters computed for the tri- and pentanickel derivatives.a
| %HFX | Kσδb | Jσ = tσ2/U | Jδ = tδ2/U | Bij | λij | Jeff | |
| Ni3(dpa)4Cl2 | 10 | 523.8 | –195.9 | –15.00 | –180.9 | 5.11 | –218.6 |
| 15 | 576.7 | –148.8 | –8.94 | –139.8 | 2.86 | –161.9 | |
| 20 | 613.6 | –117.4 | –6.13 | –111.2 | 1.73 | –126.0 | |
| 25 | 670.4 | –90.70 | –2.83 | –87.91 | 1.02 | –95.0 | |
| 30 | 712.7 | –74.1 | –0.00 | –74.12 | 0.718 | –75.0 | |
| Exp.c | –218 | ||||||
| Ni5(tpda)4Cl2 | 0 | 421.3 | –87.12 | –16.01 | –71.1 | 0.705 | –104.6 |
| 5 | 474.7 | –44.78 | –7.12 | –37.66 | 0.194 | –52.27 | |
| 10 | 525.6 | –23.80 | –3.36 | –20.40 | 0.055 | –27.26 | |
| 15 | 574.8 | –13.00 | –1.62 | –11.37 | 0.016 | –14.65 | |
| 20 | 622.1 | –7.34 | –0.76 | –6.61 | 0.0056 | –8.11 | |
| 25 | 667.2 | –4.32 | –0.42 | –3.87 | 0.0016 | –4.74 | |
| 30 | 709.7 | –2.63 | –0.24 | –2.42 | 0.0008 | –2.87 | |
| Exp.d | –33.5 |
The present analysis allows obtaining other electronic structure parameters such as the intrasite exchange integral, Kσδ, the biquadratic exchange constant, λij, and the three-body interaction constant, Bij, presented in Eqs. (2)–(5). Kσδ is an atomic parameter defined positive related to the energy difference between two parallel and antiparallel local spins. It amounts the same for both nickel compounds (at the same %HFX) as shown in Table 2. Its value decreases in parallel with the %HFX, that is, with more diffuse orbitals. As Kσδ decreases, the local triplet gradually becomes closer in energy to the open-shell singlet. In a previous report [29], we computed the rather small Kσδ = 243 meV with the B3LYP functional for the magnetic Pd(II) atoms in the hypothetical Pd3(dpa)4Cl2 compound. In that case, the smaller intrasite exchange constant associated to the diffuse 4d valence orbitals of palladium allows low-energy local singlet states.
In the case of a largely dominant tσ over tδ, the λij parameter is governed by the term (tσ)4/KU2. From Table 2, λij decreases from 5.11 to 0.718 cm−1 (10–30%HFX) in Ni3, and from 0.705 to 0.0008 (0–30%HFX) in Ni5. Thus, comparing Ni3 and Ni5 with the same functional, λij is smaller by one order of magnitude for the pentanickel compound due to its smaller tσ2/U contribution to the magnetic coupling. If we consider this parameter as the deviation from the Heisenberg model, it arises that such deviation goes to zero as the %HFX is increased, and it is much smaller in the pentanickel system. The three-body term, Bij, although being numerically larger, behaves similarly to the λij parameter. Expectedly, it increases as %HFX decreases since it is governed by the difference between Jσ and Jδ. Finally, it can be observed from the three rightmost columns of Table 2 that Jeff approaches the sum of Jσ and Jδ as Bij decreases at large amounts of HFX. Although the difference between Jeff and the two magnetic contributions is not very large in any case, it can become notable for systems with more diffuse valence orbitals, like palladate systems [29].
We have observed that the amount of HFX that comes close to reproducing the experimental values is ∼10% for the nickel complexes analyzed. In a private communication, Prof. Bénard pointed out that the recently calculated J for longer nickel chains is also smaller than expected. Thus, this behaviour for Nin string complexes could be general to systems with any n. The present data show that the B3LYP functional not always overestimates J, as generally accepted. Indeed, we report a general underestimation when a dominating Jσ coupling based on weak metal-metal interactions is present. It arises that this issue can be related to the range at which electron-electron interactions responsible for antiferromagnetism take place. In the present nickel compounds, the two coupling pathways, the δ one occurring via covalent Ni-dpa bonds and the weak, non-covalent one via Ni···Ni interactions (dNi-Ni = 2.43 Å) might be very differently treated with a functional like B3LYP. We tentatively propose that range-separated functionals can help understanding this problem and reproducing the experimental magnetic coupling constants, if they get to increase the presently underestimated Jσ and decrease the overestimated Jδ for the same molecule, Ni3(dpa)4Cl2, a fact not reported before. Results obtained with the range-separated LC-ωPBE [51] functional on the trinickel compound using the procedure herein discussed give Jσ + Jδ = –155.9 cm−1, with Jσ = –152.6 cm−1 and Jδ = –3.3 cm−1. Although the total J is still underestimated, this result follows the desired trend and confirms that the main source of error in the calculation of magnetic coupling constants in S = 1 systems comes from the poor description of the σ pathway.
4 Conclusion
The theoretical analysis of [Cu3(dpa)4Cl2]+, Ni3(dpa)4Cl2 and Ni5(tpda)4Cl2 using a general spin Hamiltonian reveals that decreasing the amount of Hartree-Fock exchange in a hybrid functional produces a generalized increment of |J|. A theoretical decomposition of the total J = Jσ + Jδ has been carried out to analyze the total J in tri- and pentanickel complexes. The previously reported DFT/B3LYP underestimation of J originates in the dominant Jσ component since it is poorly described with this functional. On the other hand, we assume that Jδ is overestimated in both nickel complexes by generalization of the findings made for [Cu3(dpa)4Cl2]+ in the present and previous reports. For the Ni5(tpda)4Cl2 complex, B3LYP also underestimates the total J with respect to the experimental value, thus exhibiting the same trend as its trinuclear congener. We suggest that B3LYP fails to reproduce the weak overlap between the neighbouring dz2-like atomic orbitals responsible for the σ interaction. From this study, we learnt (i) the uniqueness of nickel complexes of the EMAC family from the magnetic point of view due to the presence of a dominating Jσ coupling based on weak metal-metal interactions, and (ii) that the B3LYP functional not always overestimates J, as generally accepted, but it can actually underestimate it when the range at which electron-electron interactions responsible for antiferromagnetism does not take place via metal-ligand covalent bonds. In line with the last possibility, we observed that Jσ grows and Jδ decreases if a range-separated LC-ωPBE functional is used.
Acknowledgements
This work was supported by the Spanish MICINN (CTQ2008 – 06644-C02-01/BQU) and by the Generalitat de Catalunya (2009SGR-00462). X.L. thanks the Ramón y Cajal program (RYC-2008-02493).
1 For Ni5(tpda)4Cl2, the exchange and hopping integrals (K and tδ) were computed with the T2 energy rather than S2. This is because S2 is much higher in energy for this system, and the error associated with the Q–S2 gap is unacceptably large yielding an imaginary tδ. The use of T2 to calculate tδ is more reliable since the Q–T2 gap is about half of Q–S2.
2 This fact cannot be tested because there is no way to decompose both magnetic contributions experimentally.