

1 Introduction
The chemistry of organosilicon compounds can provide a wealth of chemical and biological diversity for medicinal drugs design. The incorporation of silicon atom into a drug structures has been used to improve pharmacological potency, to modify selectivity toward a given target, to change metabolic rates, and specifically to increase lipophilicity. A small increase in lipophilicity can provide several physiological benefits, including increased bioavailability as well as tissue and cell penetration [1–3]. Some of silicon-containing drug structures such as Tac101 and BNP1350 have entered human clinical trials for the treatment of cancer [2] (Fig. 1).
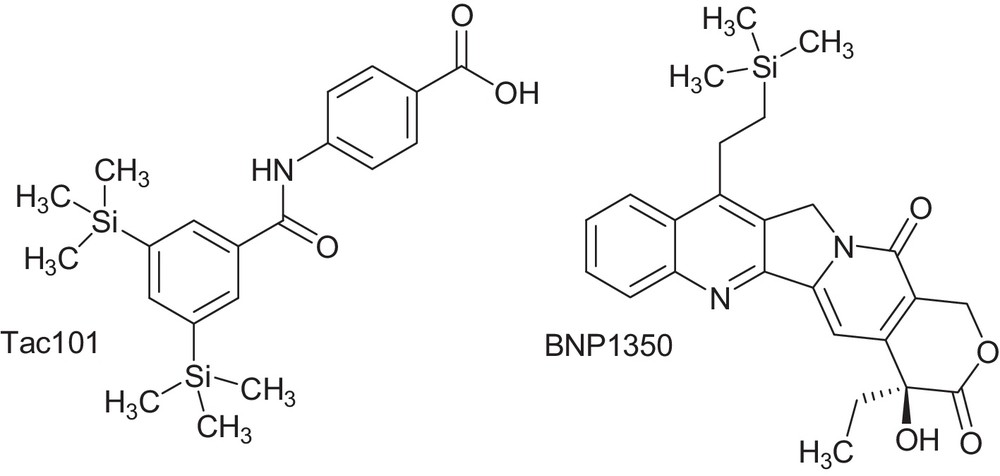
Potent silicon-based drugs.
The imidazole core constitutes an active backbone in exciting medications and is present in natural products, e.g., Losartan, Olmesartan, Eprosartan, Pimobendan, Trifenagrel, and Naamidine A (Fig. 2). The potency and wide applicability of the imidazole pharmacophore can be attributed to its hydrogen bond donor–acceptor capability as well as its high affinity for metals that are present in many protein active sites. Some of imidazole derivatives could be used as ionic liquids, anion sensors, electrical and optical materials, in molecular switches and organic light-emitting diodes (OLEDs). This versatile applicability highlights the importance of access to efficient synthetic routes to well benign fully substituted imidazoles [4–9]. Cyclizations via one-pot multicomponent coupling reactions (MCRs) are widely used for eco-compatible syntheses [10].
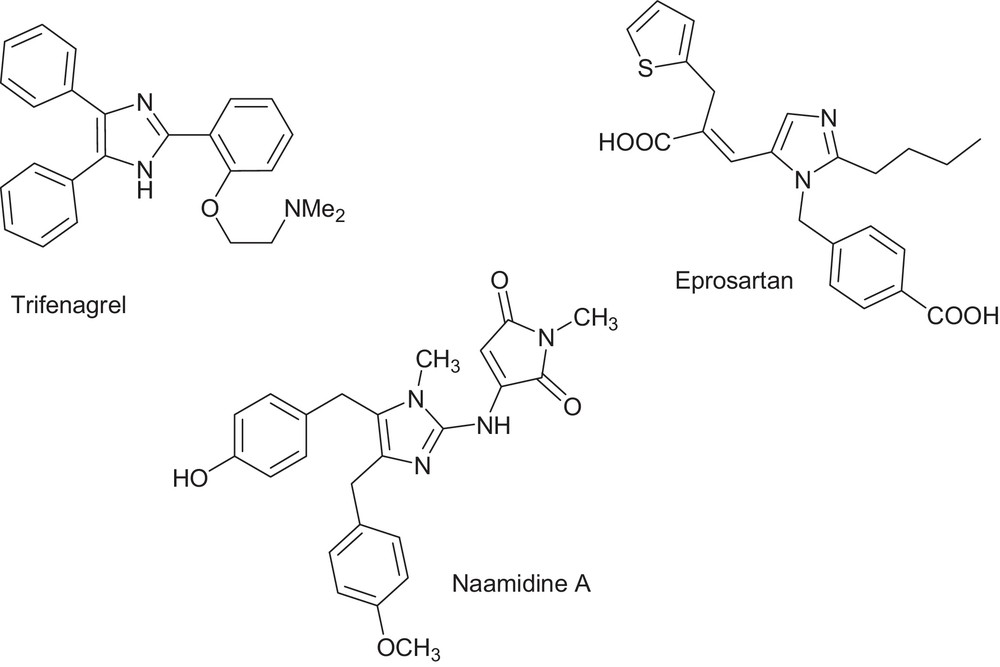
Potent imidazole-based drugs.
Multisubstituted imidazoles are generally synthesized by one-step condensation of an aldehyde, benzil, ammonium acetate and primary aromatic amine in the presence of various catalysts and green techniques [8,11–13]. Now, the use of ultrasound irradiation in different areas of chemistry has grabbed significant levels, not only for the possibility to perform environmentally benign syntheses, but also for the excellent yields they allow [14–18]. Many organic reactions have been devised, in which reagents are supported on various inorganic solid supports [19]. Various metal-supported zeolites, such as Cu-modified zeolites (HY, H–USY, H–Beta, Mordenite and ZSM-5), have been given more attention for catalytic applications [20]. Recently, silicoaluminophosphate (SAPO-n) zeolites, as small pore-sized molecular sieves, have been successfully used by several industrial researchers [21–23]. No reports are available on the synthesis of fully substituted imidazoles by use of H-SAPO-34 zeolites. Therefore, investigation of the preparation of tetraaryl imidazoles using M/SAPO-34 (M: Fe, Co, Mn, and Cu) zeolite catalysts as heterogeneous “E” catalysts (efficient, eco-friendly and economic) via zeolite-catalyzed multicomponent reactions (ZCMCRs) [24] was carried out in this study. In continuation of our interest in the synthesis of useful heterocyclic compounds possessing an imidazole nucleus and because of the importance of organosilicon compounds, we have synthesized a series of new, multi-substituted imidazoles containing organosilyl groups including carbosilanes (Si–C) and silyl ethers (Si–O).
2 Results and discussion
The catalytic influence of the different transition metals supported on the H-SAPO-34 zeolite, of the weight loadings of metal (wt.%), of the amount of catalysts has been studied here with different reaction media that were tested for the preparation of highly substituted imidazole derivatives. A one-pot four-component condensation of benzil (1 mmol), benzaldehyde (1 mmol), p-methylaniline (1 mmol), and ammonium acetate (1.1 mmol) was used as a model reaction for the synthesis of 2,4,5-triphenyl-1-p-tolyl-1H-imidazole 1a in water under ultrasound irradiation (high intensity). In the absence of a catalyst, the reaction was incomplete, even after 2 h of sonication, though the formation of a small amount of 1a (20%) was observed. When the model reaction was carried out in the presence of an H-SAPO-34 support (10 wt.%) in the same conditions, the product 1a was isolated with 32% yield, after 60 min. However, the reaction proceeded rather slowly and no significant acceleration was observed. We showed that the incorporation of different transition metals into the H-SAPO-34 support improves the cyclo-condensation reaction leading to the synthesis of a series of M/SAPO-34 (M: Fe, Co, Mn, and Cu) catalysts. All catalysts showed good yield and only produced trace amounts of by-product 2,4,5-triphenyl-1H-imidazole, at Mn-based catalyst. Metal loading on the support was done at two different loading of 5.0 and 10 wt.% of metal on the support. The results showed that the activity of catalysts with a metal loading of 10 wt.% is lower with a 5 wt.% loading. This decrease can be attributed to excessive metal agglomeration, leading to the formation of large metal particles. In addition, further content of metal blocks the pores and the active sites of the catalyst so that catalytic activity decreases. The best results (95% yield, after 3 min) were obtained in the presence of 5 wt.% of Cu/SAPO-34 in aqueous medium under ultrasound irradiation (Table 1, entry 1). This result is in agreement with our working hypothesis that the copper sites, acidity and reactant adsorption ability of the support might be the main factors allowing one to improve the cyclo-condensation reaction on Cu(NO3)2/SAPO-34.
Optimization of the reaction conditionsa.
| Entry | Catalyst (wt.%) | Time (min) | Yield (%)b |
| 1 | Cu/SAPO-34 (10) | 30 | 78 |
| 2 | Cu/SAPO-34 (5) | 5 | 95 |
| 3 | Fe/SAPO-34 (10) | 30 | 65 |
| 4 | Fe/SAPO-34 (5) | 5 | 71 |
| 5 | Co/SAPO-34 (10) | 30 | 63 |
| 6 | Co/SAPO-34 (5) | 5 | 68 |
| 7 | Mn/SAPO-34 (10) | 30 | 48 |
| 8 | Mn/SAPO-34 (5) | 5 | 50 |
| 9 | HSAPO-34 (10) | 60 | 32 |
| 10 | – | 120 | 20 |
a Reaction conditions: benzaldehyde (1.0 mmol), benzil (1.0 mmol), 4-methylaniline (1.0 mmol), ammonium acetate (1.1 mmol), and 5 wt.% Cu/SAPO-34 in water under ultrasonic irradiation at ambient temperature at 20% of the processor's power. (The metal content was kept at 5 wt.% for M/SAPO-34 catalysts).
b The indicated yields refer to isolated products.
It is also worthy to note that ultrasound-assisted reactions proceed by an acoustic cavitation phenomenon, i.e. the formation, growth, and collapse of bubbles in the liquid medium. During the collapse of a cavity, high local temperatures and pressures arise, which lead to an increase in the rate of reactions [14–17]. When other solvents such as methanol, acetonitrile, ethanol, THF, and ethylacetate were used, the reactions were slower and the yields lower.
The reactions of various aldehydes, benzil, amines, and ammonium acetate were examined in the presence of Cu/SAPO-34 (5 wt.%) in water under ultrasound irradiation. In all cases, the requisite imidazole derivatives were obtained within 3 to 5 min in excellent yields (Table 2) without the formation of any side products such as 2,4,5-trisubstituted imidazoles, oxidized products of anilines, and aldehydes, which are normally observed under the influence of strong acids. Some of the tetraaryl imidazoles are known compounds and their identity was confirmed by IR, NMR, elemental analyses, and melting points.
Synthesis of 1,2,4,5-tetraaryl imidazoles catalyzed by Cu/SAPO-34 nanocatalyst.
| Entry | Product | R1 | R2 | Time (min) | Yield (%)a | MP (°c) | |
| Found | Report | ||||||
| 1 | 1a | C6H5 | 4-MeC6H4 | 5 | 95 | 183–184 | 183–185 [32] |
| 2 | 1b | 4-MeC6H4 | C6H5 | 7 | 85 | 183–185 | 182–184 [33] |
| 3 | 1c | 4-OMeC6H4 | C6H5 | 4 | 90 | 184–186 | 186 [13] |
| 4 | 1d | 4-MeC6H4 | 4-FC6H4 | 7 | 85 | 188–190 | 188–190 [31] |
| 5 | 1e | 4-ClC6H4 | 4-MeC6H4 | 4 | 96 | 167–169 | 167–169 [31] |
| 6 | 1f | 4-MeC6H4 | 4-ClC6H4 | 7 | 84 | 166–168 | 167–169 [34] |
| 7 | 1g | 4-MeC6H4 | 3-ClC6H4 | 7 | 85 | 198–200 | 198–200 [31] |
| 8 | 1h | C6H5 | 4-OHC6H4 | 5 | 85 | > 280 | > 280 [35] |
| 9 | 1i | 4-OHC6H4 | 4-OHC6H4 | 5 | 82 | > 280 | > 280 [35] |
| 10 | 1j | 4-ClC6H4 | 4-OHC6H4 | 4 | 92 | > 280 | > 280 [35] |
| 11 | 1k | 4-OHC6H4 | 4-MeC6H4 | 8 | 83 | > 280 | 298–300 |
| 12 | 1l | 4-MeC6H4 | 4-BrC6H4 | 7 | 84 | 190–192 | 190–192 [31] |
| 13 | 1 m | 4-MeC6H4 | 4-MeC6H4 | 8 | 80 | 189–191 | 189–191 [34] |
| 14 | 1n | 4-ClC6H4 | C6H5 | 5 | 88 | 152–155 | 154 [13] |
| 15 | 1o | 2-C4H3S | 4-MeC6H4 | 5 | 88 | 177–179 | – |
| 16 | 1p | 4-MeC6H4 | C7H7 | 7 | 87 | 180–182 | – |
a Indicated yields refer to isolated products.
A plausible mechanism for the catalytic activity of Cu/SAPO-34 is postulated in Scheme 1. Cu/SAPO-34 activates the aldehyde's carbonyl group in order to facilitate the formation of diamine intermediate A. This catalyst also activates the diketone to make easier the condensation with intermediate A, to give imidazol-5-ol intermediate C that, upon elimination of water, is transformed into the desired 1,2,4,5-tetraaryl imidazole.
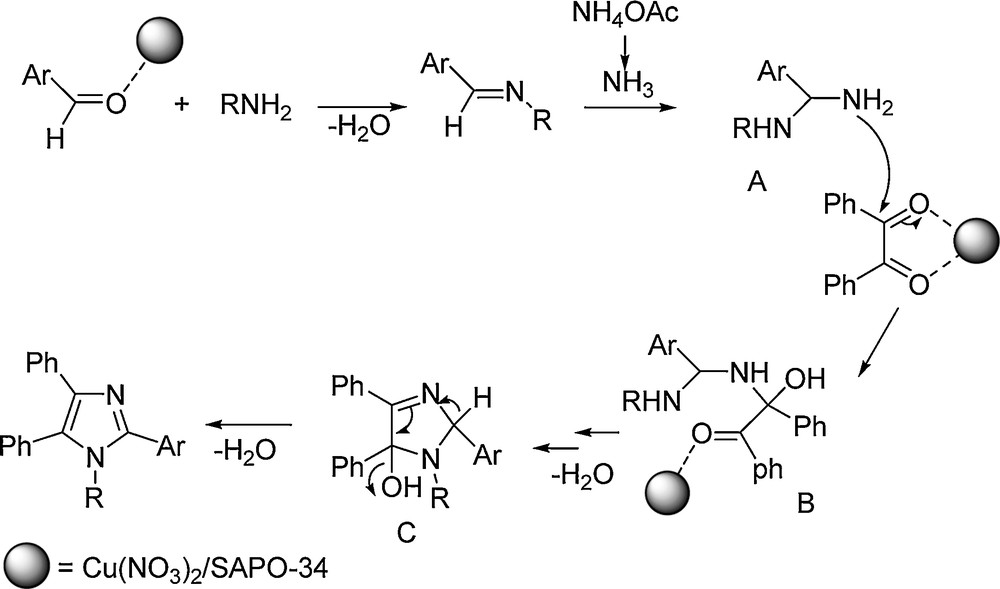
Plausible mechanism for the formation of 1,2,4,5-tetraaryl imidazoles in the presence of Cu(NO3)2/SAPO-34.
We have recently used bulky tris(trimethylsilyl)methyllithium (TsiLi) as a reagent for the preparation of vinylsilanes, epoxysilanes, halovinylsilanes, silyl ethers, etc. [25–30]. In the previous publication, we reported the synthesis of bulky organosilicon groups containing highly substituted imidazoles using silyl-substituted organometallic reagents (RSiMe2)3CLi (R = H, Me, Ph) [31,36]. In continuation of our research in this field, the new organosilyl derivatives of imidazole 1b were synthesized by using its lithium derivative, which was prepared via the metalation of 1b with lithiumdiisopropylamide (LDA) in THF, at room temperature. Trapping of this active intermediate by dropping silyl chlorides such as dimethylsilylchloride, trimethylsilylchloride, triethylsilylchloride, and triphenylsilylchloride resulted in the desired products (Scheme 2).
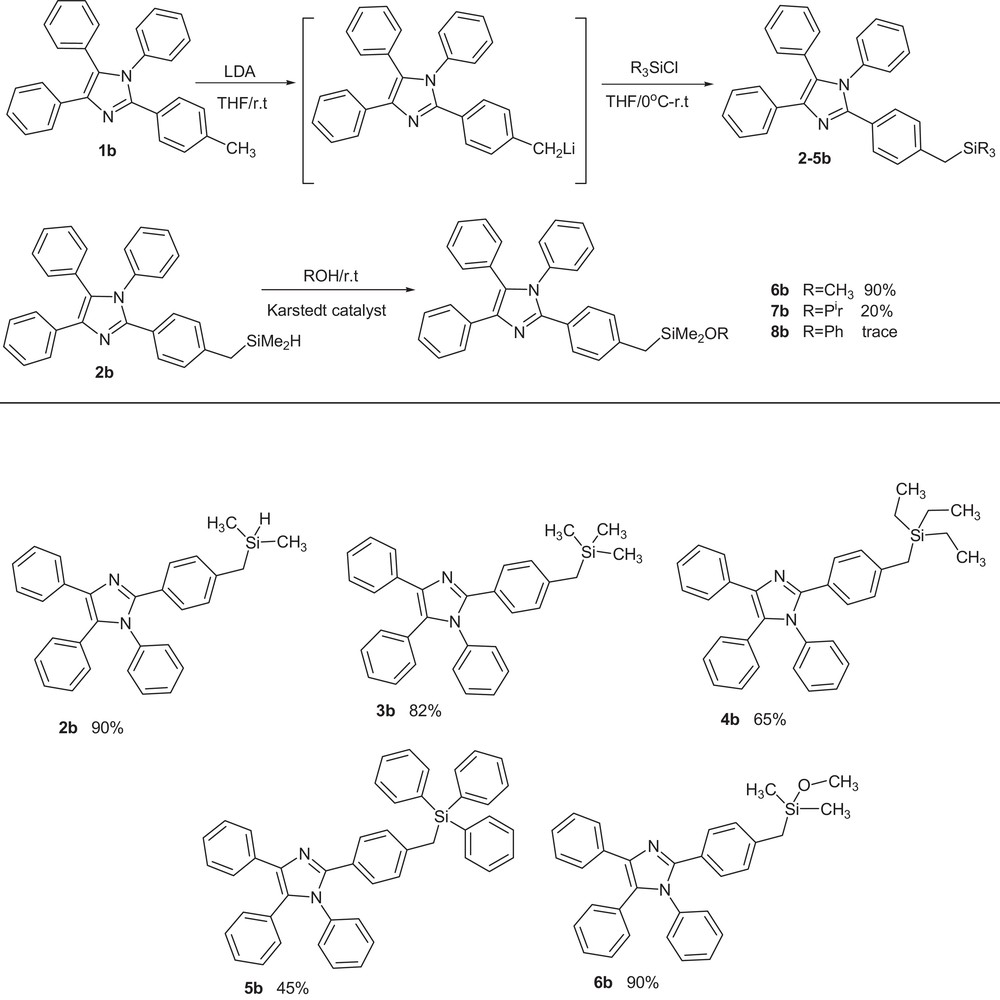
Synthesis of 1,2,4,5-tetraaryl imidazoles containing organosilyl groups.
In contrast, the reaction of silyl chloride derivatives with organolithium reagent prepared via the metalation of 1a proceeded slowly; after 2 h, it gave a poor yield. Imidazole 2b having an hydrosilane (Si–H) substituent is potentially useful for the preparation of new derivatives of imidazole containing organosilicon groups (silyl ethers) via the hydrocoupling reactions between compound 2b and some alcohols under Karstedt's catalyst (platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex, solution in xylene). Reaction of 2b with methanol as a primary alcohol gave higher yields than the analogous reaction with secondary alcohols such as isopropyl alcohol and phenol, probably because of a steric hindrance increase.
3 Conclusion
In this work, Cu(II) nitrate impregnated H-SAPO-34 zeolite has been used as an efficient supported reagent for an improved and rapid one-pot synthesis of 1,2,4,5-tetraaryl imidazoles in water under ultrasound irradiation in excellent yields. This simple procedure combined with easy recovery and reuse of the catalyst makes this method economically and environmentally benign. Some of imidazole derivatives were studied for the synthesis of a series of new, multi-substituted imidazoles containing organosilyl groups including carbosilanes (Si–C) and silyl ethers (Si–O). The synthetic intermediate 2-(4-((methoxydimethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole 6b is a potential precursor compound for the synthesis of other tetraaryl imidazole-substituted (alkoxydimethylsilyl)methanes through the transetherification reactions 6b with other alcohols and polymers bearing hydroxyl groups. We shall investigate this possibility and discuss our results in future communications.
4 Experimental
4.1 Material and technique
Chemicals were either prepared in our laboratory or purchased from Merck, Fluka, and Aldrich. Commercial products were used without further purification. The 1H NMR and 13C NMR spectra were recorded with a Bruker FT-400 MHz spectrometer at room temperature and with CDCl3 and/or DMSO-d6 as a solvent. The FT-IR spectra were recorded on a Bruker Tensor 270 spectrometer. The reactions were carried out by used of an ultrasonic processor probe (SONOPULS Ultrasonic homogenizers). The Elemental analyses were carried out with an Elementar vario EL III instrument. The abbreviations used for NMR signals are: s = singlet, d = doublet, t = triplet, and m = multiplet. Melting points were recorded on a Büchi B-545 apparatus in open capillary tubes and are uncorrected.
4.2 Nanocatalysts preparation
Catalysts were prepared by the homogeneous deposition precipitation (HDP) method. In the HDP method, an aqueous solution of urea is utilized to generate ammonium hydroxide as a precipitating agent at elevated temperature, according to the following reaction: CO(NH2)2 + 3H2O → CO2 + 2NH4+ + 2OH−.
The precursors of the used metals were Co(NO3)2·6H2O, Fe(NO3)3·9H2O, Cu(NO3)2·3H2O, and Mn(NO3)2·4H2O, and the support used was SAPO-34. Metal loading on a support was done at two different metal loadings of 5.0 and 10 wt%. In a typical procedure, the SAPO-34 support was added to a solution containing an appropriate amount of metal nitrate under vigorous stirring, and then urea was added to the mixture. The temperature was gradually increased to 95 °C and maintained at that value for 5 h until the hydroxide precipitate was complete. Thereafter, the mixture was aged at room temperature for 14 h and then the solid was filtered, washed thoroughly with deionized water, air dried at 90 °C for 14 h and then finally calcined at 550 °C for 4 h [36].
4.3 General procedure for the preparation of 1,2,4,5-tetraaryl imidazoles (1a–1p)
A mixture of benzil (1 mmol), aldehyde (1 mmol), amine (1 mmol), and ammonium acetate (1.1 mmol) as ammonia source and 5 wt.% of Cu/SAPO-34 in 3 of water was introduced in a 20-ml heavy walled pear-shaped two-necked flask with a nonstandard tapered outer joint. The flask was attached to a 12-mm tip diameter probe and the reaction mixture was sonicated at ambient temperature at 20% of the processor's power. After completion of the reaction (monitored by TLC, within 2–6 min), the solid product was filtered, washed with water, dried, and recrystallized from ethanol. The supported reagent was washed thrice with water and ethanol and dried under vacuum before reuse.
4.3.1 Spectral data of some of the selected products
4.3.1.1 1-(4-Fluorophenyl)-4,5-diphenyl-2-p-tolyl-1H-imidazole (1d)
White powder; yield 97%; m.p. = 188–190 °C; FTIR (KBr, cm−1): 3058 (Ar–H), 2962 (C–H), 1602, 1508, 1444 (C = C), 960, 775, 730. 1H NMR (400 MHz, CDCl3, ppm): δ 2.36 (s, 3H, Ar–CH3), 6.96–7.00 (m, 2H, Ar–H), 7.03–7.07 (m, 2H, Ar–H), 7.11 (d, 2H, J = 8.0 Hz, Ar–H), 7.14–7.17 (m, 2H, Ar–H), 7.21–7.32 (m, 6H, Ar–H). 7.35 (d, 2H, J = 8.0 Hz, Ar–H), 7.63 (d, 2H, J = 7.2 Hz, Ar–H), 13C NMR (100 MHz, CDCl3, ppm): δ 20.24 (Ar–CH3), 114.95, 115.17, 125.58, 126.32, 126.41, 127.01, 127.12, 127.41, 127.79, 127.88, 128.99, 129.08, 129.50, 129.58, 130.07, 132.19, 133.30, 137.16, 137.31, 146.12, 159.59, 162.06 (Ar). Anal. calcd. for C28H21FN2: C 83.14, H 5.23, N 6.93%. Found: C 83.09, H 5.14, N 6.81%.
4.3.1.2 2-(4-Chlorophenyl)-4,5-diphenyl-1-p-tolyl-1H-imidazole (1e)
White powder; yield 96%; m.p. = 167–169 °C; FTIR (KBr, cm−1): 3054 (Ar–H), 2976 (C–H), 1600, 1510, 1478, 1445 (C = C), 745, 696. 1H NMR (400 MHz, CDCl3, ppm): δ 2.36 (s, 3H, Ar–CH3), 6.95 (d, J = 8.1 Hz, 2H, Ar–H), 7.10 (d, J = 8.1 Hz, 2H, Ar–H), 7.16–7.18 (m, 2H, Ar–H), 7.21–7.30 (m, 8H, Ar–H), 7.41–7.43 (m, 2H, Ar–H), 7.61–7.63 (m, 2H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ 20.14 (Ar–CH3), 119.77, 125.61, 126.30, 126.95, 126.99, 127.13, 127.29, 127.31, 128.10, 128.82, 129.03, 129.50, 130.05, 130.17, 133.15, 133.21, 133.31, 137.41, 144.74 (Ar). Anal. calcd. for C28H21ClN2: C 79.89, H 5.03, N 6.66%. Found: C 79.76, H 5.14, N 6.86%.
4.3.1.3 1-(3-Chlorophenyl)-4,5-diphenyl-2-p-tolyl-1H-imidazole (1g)
White powder; yield 94%; m.p. = 198–200 °C; FTIR (KBr, cm−1): 3056 (Ar–H), 2963 (C–H), 1601, 1490, 1479 (C = C), 739, 703. 1H NMR (400 MHz, CDCl3, ppm): δ 2.33 (s, 3H, Ar–CH3), 6.93-6.95 (m, 1H, Ar–H), 7.05 (t, J = 1.86 Hz, 1H, Ar–H), 7.08 (d, 2H, J = 8.04 Hz, Ar–H), 7.12–7.27 (m, 10H, Ar–H), 7.32 (d, 2H, J = 8.15 Hz, Ar–H),7.58–7.60 (m, 2H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ 20.27 (Ar–CH3), 125.64, 125.77, 126.24, 126.33, 127.14, 127.15, 127.47, 127.58, 127.78, 127.95, 128.92, 129.31, 130.05, 133.21, 133.48, 137.33, 137.45, 146.00 (Ar). Anal. calcd. for C28H21ClN2: C 79.89, H 5.03, N 6.66%. Found: C 79.78, H 5.12, N 6.74%.
4.3.1.4 4-(2,4,5-Triphenyl-1H-imidazol-1-yl)phenol (1h)
White powder; yield 92%; m.p. >280 °C; FTIR (KBr, cm−1): 3414 (OH), 3054 (Ar–H), 2922 (C–H), 1604, 1513, 1446 (C = C), 696. 1H NMR (400 MHz, CDCl3, ppm): δ 6.66 (d, J = 8.5 Hz, 2H, Ar–H), 7.04 (d, J = 8.5 Hz, 2H, Ar–H), 7.14–7.30 (m, 11H, Ar–H), 7.42–7.44 (m, 2H, Ar–H), 7.49 (d, J = 7.5 Hz, 2H, Ar–H), 9.78 (s, 1H, Ar–OH). 13C NMR (100 MHz, CDCl3, ppm): δ 115.59, 126.35, 127.84, 128.12, 128.15, 128.27, 128.40, 129.79, 130.58, 130.63, 131.12, 131.59, 134.56, 136.61, 146.11, 157.31 (Ar). Anal. calcd. for C27H20N2O: C 83.48, H 5.19, N 7.21%. Found: C 83.54, H 5.07, N 7.19%.
4.3.1.5 4,4′-(4,5-Diphenyl-1H-imidazole-1,2-diyl)diphenol (1i)
White powder; yield 91%; m.p. >280 °C; FTIR (KBr, cm−1): 3416 (OH), 3053 (Ar–H), 2924 (C–H), 1642, 1513, 1438 (C = C), 694. 1H NMR (400 MHz, CDCl3, ppm): δ 6.64–6.67 (m, 4H, Ar–H), 7.00 (d, J = 8.4 Hz, 2H, Ar–H), 7.12–7.29 (m, 10H, Ar–H), 7.45 (d, J = 7.7 Hz, 2H, Ar–H), 9.73 (bs, 2H, Ar–OH). 13C NMR (100 MHz, CDCl3, ppm): δ 114.92, 115.55, 121.48, 126.22, 126.31, 128.05, 128.09, 128.16, 128.39, 129.67, 129.84, 130.86, 130.89, 131.14, 134.75, 136.15, 146.56, 157.19, 157.52 (Ar). Anal. calcd. for C27H20N2O2: C 80.18, H 4.98, N 6.93%. Found: C 80.01, H 4.75, N 6.87%.
4.3.1.6 4-(2-(4-Chlorophenyl)-4,5-diphenyl-1H-imidazol-1-yl)phenol (1j)
White powder; yield 94%; m.p. >280 °C; FTIR (KBr, cm−1): 3416 (OH), 3054 (Ar–H), 2926 (C–H), 1595, 1513, 1478, 1440 (C = C), 745, 695. 1H NMR (400 MHz, CDCl3, ppm): δ 6.67 (d, J = 8.4 Hz, 2H, Ar–H), 7.05 (d, J = 8.4 Hz, 2H, Ar–H), 7.14–7.43 (m, 12H, Ar–H), 7.47 (d, J = 7.5 Hz, 2H, Ar–H), 9.82 (s, 1H, Ar–OH). 13C NMR (100 MHz, CDCl3, ppm): δ 115.69, 126.33, 126.46, 127.55, 128.15, 128.27, 128.37, 128.43, 129.37, 129.71, 129.74, 130.44, 131.10, 131.89, 132.98, 134.37, 136.76, 144.96, 157.42 (Ar). Anal. calcd. for C27H19ClN2O: C 76.68, H 4.53, N 6.62%. Found: C 76.49, H 4.61, N 6.58%.
4.3.1.7 1-(4-Bromophenyl)-4,5-diphenyl-2-p-tolyl-1H-imidazole (1l)
White powder; yield 90%; m.p. = 190–192 °C; FTIR (KBr, cm−1): 3057 (Ar–H), 2961 (C–H), 1602, 1570, 1510, 1478 (C = C), 726, 697. 1H NMR (400 MHz, CDCl3, ppm): δ 2.36 (s, 3H, Ar–CH3), 6.93 (d, J = 8.6 Hz, 2H, Ar–H), 7.11–7.17 (m, 4H, Ar–H), 7.20–7.35 (m, 8H, Ar–H), 7.41 (d, 2H, J = 8.6 Hz, Ar–H), 7.60–7.62 (m, 2H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ 20.27 (Ar–CH3), 121.06, 121.58, 125.64, 126.37, 127.14, 127.51, 127.88, 127.96, 128.56, 128.91, 129.35, 129.44, 130.10, 131.13, 131.25, 133.27, 135.27, 137.42, 146.04 (Ar). Anal. calcd. for C28H21BrN2: C 72.26, H 4.55, N 6.02%. Found: C 72.14, H 4.47, N 6.14%.
4.3.1.8 4,5-Diphenyl-2-(thiophen-2-yl)-1-p-tolyl-1H-imidazole (1o)
Pink powder; yield 91%; m.p. = 178–180 °C; FTIR (KBr, cm−1): 3060 (Ar–H), 2930 (C–H), 1603, 1490, 1448 (C = C), 1398, 1237, 1115, 1072, 1026, 968, 917, 771, 699. 1H NMR (400 MHz, CDCl3, ppm): δ 2.41 (s, 3H, Ar–CH3), 6.73 (d, J = 3.4 Hz, 1H, Ar–H), 6.90 (t, J = 4.4 Hz, 1H, Ar–H), 7.12 (d, J = 8.2 Hz, 2H, Ar–H), 7.18–7.30 (m, 11H, Ar–H), 7.62 (d, J = 7.2 Hz, 2H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ 20.29 (Ar–CH3), 125.08, 125.20, 125.58, 126.09, 126.32, 126.93, 127.11, 127.30, 127.64, 128.95, 129.39, 130.01, 132.28, 132.95, 133.26, 137.21, 138.16 (Ar). Anal. calcd. for C26H20N2S: C 79.56, H 5.14, N 7.14, S 8.17%. Found: C 79.38, H 5.25, N 7.01, S 8.31%.
4.3.1.9 1-Benzyl-4,5-diphenyl-2-p-tolyl-1H-imidazole (1p)
White powder; yield 90%; m.p. = 180–182 °C; FTIR (KBr, cm−1): 3031 (Ar–H), 2918 (C–H), 1619, 1509, 1478, 1442 (C = C), 934, 773, 698. 1H NMR (400 MHz, CDCl3, ppm): δ 2.41 (s, 3H, Ar–CH3), 5.14 (s, 2H, CH2), 6.84–6.86 (m, 2H, Ar–H), 7.18 (d, J = 7.1 Hz, 1H, Ar–H), 7.23–7.44 (m, 12H, Ar–H), 7.58–7.63 (m, 4H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ 20.30 (Ar–CH3), 47.22 (CH2), 124.97, 125.24, 125.74, 126.25, 127.01, 127.50, 127.70, 127.74, 127.84, 127.90, 128.03, 128.84, 130.05, 130.12, 136.64, 137.76 (Ar). Anal. calcd. for C29H24N2: C 86.97, H 6.04, N 6.99%. Found: C 86.81, H 6.20, N 7.02%.
4.4 Procedure for synthesis of 2-(4-((dimethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (2b) and 2-(4-((trimethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (3b)
To a stirred THF solution (15 ml) of diisopropylamine (20 mmol), a cyclohexane solution (3 ml) of n-butyllithium (0.65 g, 10 mmol) was added in a 100-ml round-bottomed flask equipped with a three-way stop-cock and an isobaric-type dropping funnel. After stirring for a few minutes to complete the formation of lithium diisopropylamide (LDA), 1,2,4-triphenyl-5-p-tolyl-1H-imidazole (1b) (10 mmol) was added. The color of the mixture turned brown immediately. Then an excess amount of dimethylsilyl chloride or trimethylsilyl chloride (36 mmol) in THF (15 ml) was dropped from the isobaric-type dropping funnel into the mixture for 1 h at 0 °C. After completion of the reaction, low-temperature-boiling materials were evaporated and the residue was chromatographed over silica gel by 10:1 n-hexane:ethyl acetate to yield 90% of (2b) and 82% of (3b).
4.4.1 The spectral data of selected products
4.4.1.1 2-(4-((Dimethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (2b)
White powder; yield 90%, m.p. = 180–182 °C; FTIR (KBr, cm−1): 3055 (Ar–H), 2960 (C–H), 2922, 2113 (Si-H), 1563, 1491, 1450, 1418 (C = C), 1390, 1260, 960, 850 (Si–CH3), 764, 694. 1H NMR (400 MHz, CDCl3, ppm): δ 0.1–0.11 (d, 6H, SiMe2), 2.15 (s, 2H, CH2), 3.94–3.96 (m, 1H, S-H), 6.95 (d, J = 7.9 Hz, 2H, Ar–H), 7.07 (d, 2H, J = 7.6 Hz, Ar–H), 7.16 (d, 2H, J = 7.5 Hz, Ar–H). 7.21–7.33 (m, 11H, Ar–H), 7.64 (d, 2H, J = 7.6 Hz, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ -5.81 (SiMe2), 23.13 (CH2), 125.47, 126.36, 126.80, 127.09, 127.27, 127.40, 127.90, 127.95, 129.47, 129.70, 130.08, 133.47, 136.15, 139.44, 146.19. Anal. calcd. for C30H28N2Si: C 81.04, H 6.35, N 6.30%. Found: C 81.11, H 6.28, N 6.41%.
4.4.1.2 2-(4-((Trimethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (3b)
White powder; yield 82%, m.p. = 156–158 °C; FTIR (KBr, cm−1): 3054 (Ar–H), 2956 (C–H), 1509, 1451, 1417 (C = C), 1380, 1258, 939, 835 (Si–CH3), 768, 697. 1H NMR (400 MHz, CDCl3, ppm): δ -0.04 (s, 9H, SiMe3), 2.05 (s, 2H, CH2), 6.88 (d, J = 8.0 Hz, 2H, Ar–H), 7.03 (d, J = 6.5 Hz, 2H, Ar–H), 7.14 (d, J = 5.94 Hz, 2H, Ar–H), 7.13–7.29 (m, 11H, Ar–H) 7.62 (d, 2H, J = 7.4 Hz, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ -3.00 (SiMe3), 26.04 (CH2), 125.06, 125.47, 125.92, 126.39, 126.67, 126.82, 127.09, 127.27, 127.41, 127.56, 127.79, 127.92, 129.43, 129.71, 130.09, 136.16, 140.03. Anal. calcd. for C31H30N2Si: C 81.18, H 6.59, N 6.11%. Found: C 81.27, H 6.39, N 6.08%.
4.5 Procedure for synthesis of 2-(4-((triethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (4b) and 2-(4-((triphenylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (5b)
After preparation of organolithium-imidazole (10 mmol) in THF solution (10 ml), a solution (15 ml) of triethylsilyl chloride or triphenylsilyl chloride (11 mmol) was dropped from the isobaric-type dropping funnel into the mixture for 3 h at room temperature. After completion of the reaction, the mixture was poured into a saturated aqueous ammonium chloride solution (50 ml) and extracted with CH2Cl2 (2 × 50 ml). The organic phase was washed with water (100 ml) and dried (Na2SO4), then the residue was chromatographed over silica gel by 10:1 n-hexane:ethyl acetate to yield 65% of (4b) and 45% of (5b).
4.5.1 The spectral data of selected products
4.5.1.1 2-(4-((Triethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (4b)
White powder; yield 65%, m.p. = 90–92 °C; FTIR (KBr, cm−1): 3056 (Ar–H), 2952 (C–H), 1597, 1565, 1527, 1490, 1447 (C = C), 1380, 1240, 939, 840 (Si–C2H5), 759, 697. 1H NMR (400 MHz, CDCl3, ppm): δ 0.49 (q, J = 7.9 Hz, 6H, SiCH2-CH3), 0.90 (t, J = 7.9 Hz, 9H, SiCH2-CH3), 2.08 (s, 2H, CH2), 6.92 (d, J = 8.0 Hz, 2H, Ar–H), 7.03 (d, J = 6.8 Hz, 2H, Ar–H), 7.14 (d, J = 6.0 Hz, 2H, Ar–H), 7.18–7.29 (m, 11H, Ar–H), 7.63 (d, J = 7.4 Hz, 2H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): δ 1.80 (SiCH2-CH3), 6.23 (SiCH2-CH3), 20.56 (CH2), 124.95, 125.42, 126.35, 126.68, 126.77, 126.99, 127.04, 127.22, 127.35, 127.79, 127.86, 129.36, 129.66, 130.03, 133.43, 136.11, 140.17, 146.26. Anal. calcd. for C34H36N2Si: C 81.55, H 7.25, N 5.59%. Found: C 81.63, H 7.16, N 5.41%.
4.5.1.2 1,4,5-Triphenyl-2-(4-((triphenylsilyl)methyl)phenyl)-1H-imidazole (5b)
White powder; yield 45%, m.p. = 120–122 °C; FTIR (KBr, cm−1): 3054 (Ar–H), 2956 (C–H), 1600, 1589, 1562, 1540, 1495, 1454 (C = C), 1383, 1249, 938, 844 (Si–C), 754, 699. 1H NMR (400 MHz, CDCl3, ppm): δ 2.88 (s, 2H, CH2), 6.98 (d, J = 7.0 Hz, 2H, Ar–H), 7.03 (d, J = 7.0 Hz, 2H, Ar–H), 7.11–7.58 (m, 28H, Ar–H), 7.62 (d, J = 6.8 Hz, 2H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm): 15.22 (CH2), 124.99, 125.94, 126.15, 126.68, 127.03, 127.25, 127.30, 127.35, 127.51, 127.79, 127.87, 127.94, 128.16, 129.51, 130.10, 135.52, 136.02, 145.55. Anal. calcd. for C46H36N2Si: C 85.67, H 5.63, N 4.34%. Found: C 85.81, H 5.54, N 4.45%.
4.6 General procedure for the synthesis of tetraaryl imidazole-substituted (alkoxydimethylsilyl)methanes
A 50-ml round-bottomed two-neck flask was charged with 2b (0.50 mmol) and ROH (20 ml) under dry argon under magnetic stirring. Karstedt's catalyst ([Pt]/[Si–H]= 7.2 × 10−3) was then added and the reaction progress was monitored. Several samples were taken at different time intervals and were analyzed by infrared (FTIR) spectroscopy. The mixture was stirred at 65–70 °C until complete disappearance of the Si–H bond in the FTIR spectra. After completion of the reaction, the mixture was allowed to cool to room temperature. Then alcohol was evaporated under reduced pressure, and the residue was purified by column chromatography (silica gel, 10:1 n-hexane:ethyl acetate) to give the corresponding products.
4.6.1 Spectral data of the selected product
4.6.1.1 2-(4-((Methoxydimethylsilyl)methyl)phenyl)-1,4,5-triphenyl-1H-imidazole (6b)
White powder; yield 90%, m.p. = 156–158 °C; FTIR (KBr, cm−1): 3061 (Ar–H), 2931 (C–H), 1600, 1600, 1566, 1497, 1447 (C = C), 1419, 1394, 1370, 1256, 937, 845 (Si–C), 1085, 1025, 955 (Si–O), 726, 697. 1H NMR (400 MHz, CDCl3, ppm): δ 0.13 (s, 6H, SiMe2), 2.17 (s, 2H, CH2), 3.38 (s, 2H, O–CH2), 7.03 (dd, J = 1.3 Hz, J = 7.8 Hz, 2H, Ar–H), 7.15 (dd, J = 1.6 Hz, J = 7.7 Hz, 2H, Ar–H), 7.20–7.32 (m, 13H, Ar–H), 7.63 (d, J = 7.2 Hz, 2H, Ar–H). 13C NMR (100 MHz, CDCl3, ppm):–4.52 (SiMe2), 18.73 (CH2), 50.00 (O–CH2), 125.48, 126.47, 126.82, 126.91, 127.10, 127.26, 127.46, 127.91, 129.50, 129.67, 129.73, 130.10, 136.12, 141.84. Anal. calcd. for C31H30N2OSi: C 78.44, H 6.37, N 5.90%. Found: C 78.58, H 6.28, N 6.01%.
Acknowledgments
The authors would like to acknowledge the financial support from the University of Tabriz and the Iranian Nanotechnology Initiative.