

1 Introduction
Amido complexes of late transition metals are of incessant interest because of potential applications for catalysis, involving the formation of carbon–nitrogen bonds [1,2]. Most recent studies have been focused on such complexes containing an arylamido ligand that may stabilize the metal–amido bond by partially delocalizing the electron density on the amido nitrogen to an aryl-substituent [2]. However, such complexes with a parent–amido ligand (NH2) have been relatively little explored [1,3–5]; nevertheless, such species are of importance as intermediates in metal-catalyzed amination reactions with ammonia [6]. Although palladium is the primary metal used in homogenous catalysis, there are a few examples of parent–amido complexes with this metal [6a,7].
Monomeric amido complexes have a strong affinity to undergo substitutional dimerization or oligomerization, particularly crucial for coordinatively unsaturated complexes, yielding amido-bridged species. We have previously reported monomeric dimethylamido and arylamido complexes of Pd(II) and Pt(II) having a phenyl substituted PCP pincer ligand [8]. Herein, we report the synthesis of dimeric and monomeric parent–amido complexes of palladium(II), in which the latter employs a sterically hindered cyclohexyl derivative of PCP pincer to preclude undergoing substitutional dimerization. Thus, the monomeric amido complex displays unique reactivity towards activated acetylenes via the insertion of carboncarbon triple bonds into the PdNH2 bond to produce diastereospecific aminated vinyl palladium(II) complexes. Further reaction of the vinyl complexes with acidic phenol to liberate diastereoselective amino olefin derivatives is described in terms of absolute diastereomeric configuration of the vinyl complex along with the reactivity feature in the ligand exchange reaction involving the σ-vinylic and the σ-phenoxide in the Pd(II) complexes.
2 Results and discussion
2.1 NH2-bridged dimeric amido complex
The reaction of a THF solution of trans-[Pd(PEt3)2(Ph)(NH3)]OTf (1) with NaNH2 at ambient temperature gives a gray suspension containing a single compound of palladium(II) along with dissociated triethylphosphine, evidenced by the 31P{1H} NMR spectroscopy (Scheme 1). A pure off-white palladium(II) dimer anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (2) was obtained from a n-pentane extract of the residues resulted from the reaction mixture. The 31P{1H} NMR spectrum of 2 in d6-benzene shows a single peak at δ 20.2 indicative of a single compound in solution. In the 13C{1H} NMR spectrum, the methylene carbon directly attached to the phosphorus resonates at δ 15.61 (1J(CP) = 26.7 Hz) as a doublet. In the 1H NMR spectrum, the bridged-amide protons (μ-NH2) have been observed at δ −1.35 as a broad signal. The microanalytical data for 2 is consistent with dimeric formulation as was judged by NMR spectroscopic data.
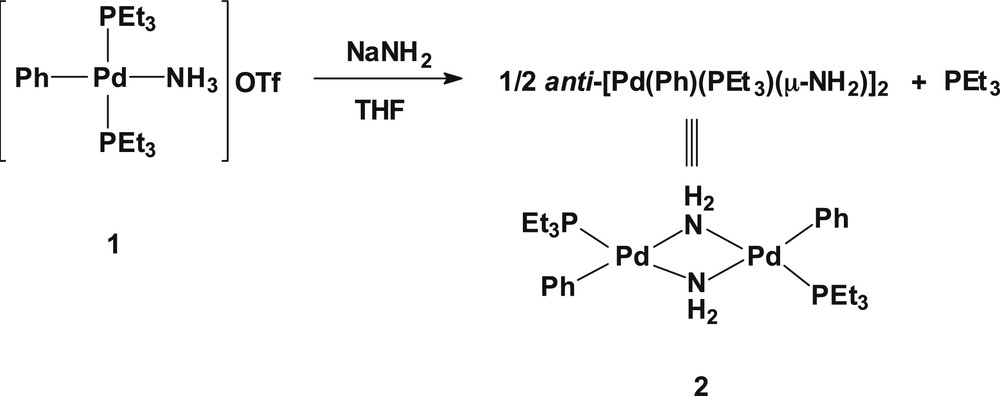
Synthesis of anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (2).
Single crystals suitable for an X-ray diffraction study were obtained by slow evaporation of an n-pentane solution of anti-[Pd(PEt3)(Ph)(μ-NH2)]2 (2). The molecular structure of one of the two crystallographically independent, but chemically identical molecules of 2 is shown in Fig. 1 [9]. The two independent molecular structures of 2 contain respective Pd2N2 rings puckered with dihedral angles of 53.1(2)° and 55.2(2)°. The Pd–Pd distances are 2.9594(10) and 2.9401(9) Å, respectively, indicating no bonding between the Pd–Pd [10], which are comparable with the Pt–Pt distances in the range of 3.087-3.134 Å found in the NH2-bridged platinum(II) dimers, anti-[Pt(POPh2)(PMePh2)(μ-NH2)]2 [4b], [Pt(PMe2Ph)2(μ-NH2)]22+ [4c], and anti-[PtMe (PPh3)(μ-NH2)]2 [5a]. The PdN bond lengths are in the range of 2.067(7)–2.127(7) Å. The PdNPd and NPdN bond angles are 88.8(3)–90.4(3)° and 77.7(3)–78.3(3)°, respectively. The dimeric complex 2 is intact to air and moisture. No reactions of 2 with unsaturated molecules such as CO2, CH2 = CHCN, dimethyl acetylenedicarboxylate (DMAD), diethyl maleate, and cyclohexene have been observed, revealing a lack of nucleophilicity on the part of the bridging NH2. Taking into account the observed stability of 2, it is rather unexpected that complex 2 is, to the best of our knowledge, the first structurally determined NH2-bridged palladium(II) dimer (precedents of structurally determined bridging arylamido complexes of palladium(II) [11]), although a few precedents of analogous platinum(II) dimers have been reported [4,5a].
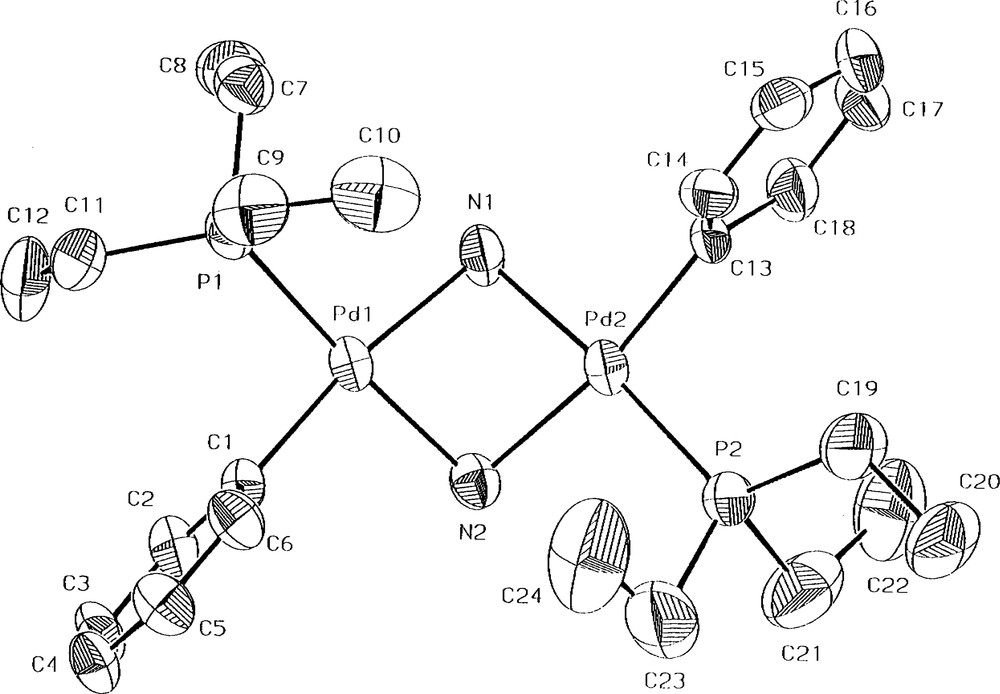
The molecular structure for one of the two crystallographically independent molecules of 2 shown with 40% thermal ellipsoids. Selected bond lengths (Å) and angles (°): Pd(1)–N(1) 2.104(7), Pd(1)–N(2) 2.085(7), Pd(2)–N(1) 2.067(7), Pd(2)–N(2) 2.127(7), Pd(1)–C(1) 2.000(9), Pd(2)–C(13) 1.998(9), Pd(1)–P(1) 2.233(3), Pd(2)–P(2) 2.235(3); Pd(2)–N(1)–Pd(1) 90.4(3), Pd(1)–N(2)–Pd(2) 89.3(3), N(2)–Pd(1)–N(1) 77.8(3), N(1)–Pd(2)–N(2) 77.7(3), C(1)–Pd(1)–N(2) 93.5(3), C(1)–Pd(1)–N(1) 171.3(3), C(1)–Pd(1)–P(1) 89.0(3), N(2)–Pd(1)–P(1) 174.3(2), N(1)–Pd(1)–P(1) 99.5(2), C(13)–Pd(2)–N(1) 92.8(3), C(13)–Pd(2)–N(2) 170.4(3), C(13)–Pd(2)–P(2) 91.7(3), N(1)–Pd(2)–P(2) 175.1(2), N(2)–Pd(2)–P(2) 97.7(2).
2.2 Monomeric amido complex
Since the formation of amide-bridged dimeric species resulted from substitutional dimerization by releasing triethylphosphine, we have used a palladium(II) ammine complex containing a trans-spanning terdentate ligand as a synthetic precursor for the preparation of a monomeric palladium(II) amide. Using the similar synthetic manner as for 2, treatment of the cationic ammine complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)](OTf) (3) with NaNH2 afforded the monomeric amido complex Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (4). When NaH was employed in place of NaNH2 as a deprotonating agent, the reaction yields a mixture of two products including mainly the complex 4 along with a small amount of palladium(II) hydride (2,6-(Cy2PCH2)2C6H3)PdH (the formation of the palladium(II) hydride (2,6-(Cy2PCH2)2C6H3)Pd–H can be verified by observation of the upfield hydride resonance at δ −3.73 t (2J(PH) = 16 Hz) in the 1H NMR spectrum [8b,12]). Alternatively, the metathetical replacement of the triflate ligand in (Pd(Cy2PCH2)2C6H3)(OTf) with NaNH2 exclusively yields the monomeric palladium(II) amide. The utilized synthetic routes are depicted in Scheme 2. The formation of Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (4) was verified by the upfield shift NH2 resonance at δ −0.20, and the relative intensity of the methylene protons in the pincer ligand and NH2 resonances in the 1H NMR spectrum. On dilution of a d6-benzene solution of 4, the NH2 resonance shifts upfield, implicating an intermolecular hydrogen bonding in the complexes. Complex 4 is highly air and moisture sensitive. When the complex is exposed to a moisturized atmosphere or trace amounts of water, it immediately protonates to convert into the cationic ammine species [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)]+, which was established by the observation of the identical 31P{1H} NMR resonance with complex 3 at δ 52.7 in d6-benzene (Scheme 3). A d6-benzene solution of 4 reacts with diphenyliodonium triflate ([Ph2I]OTf) to give the N-phenylated amine complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH2Ph)]OTf (5) (Scheme 3). The formation of the cationic aniline complex 5 can be verified by its independent preparation from the reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) with NH2Ph in d6-benzene (see Experimental section).
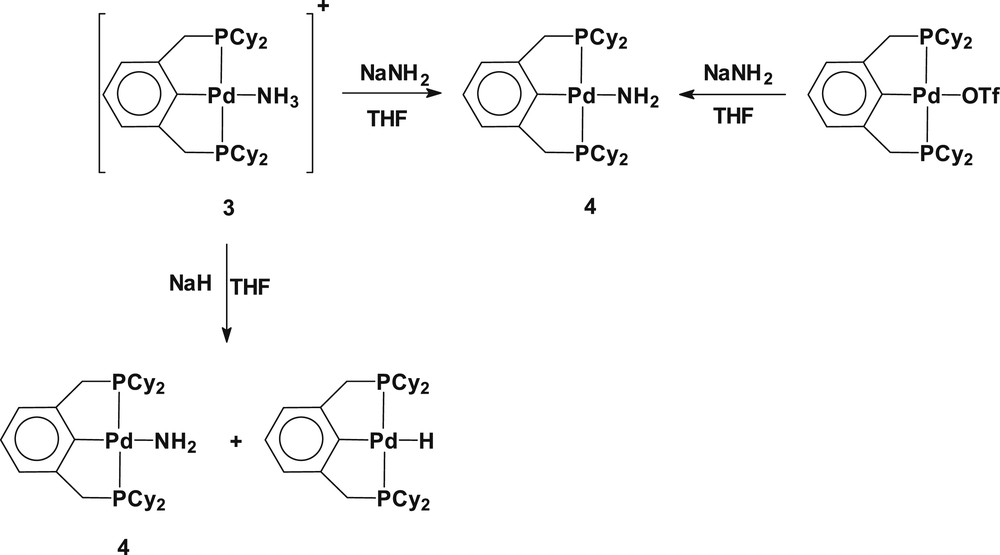
Synthesis of (2,6–(Cy2PCH2)2C6H3)Pd(NH2) (4).
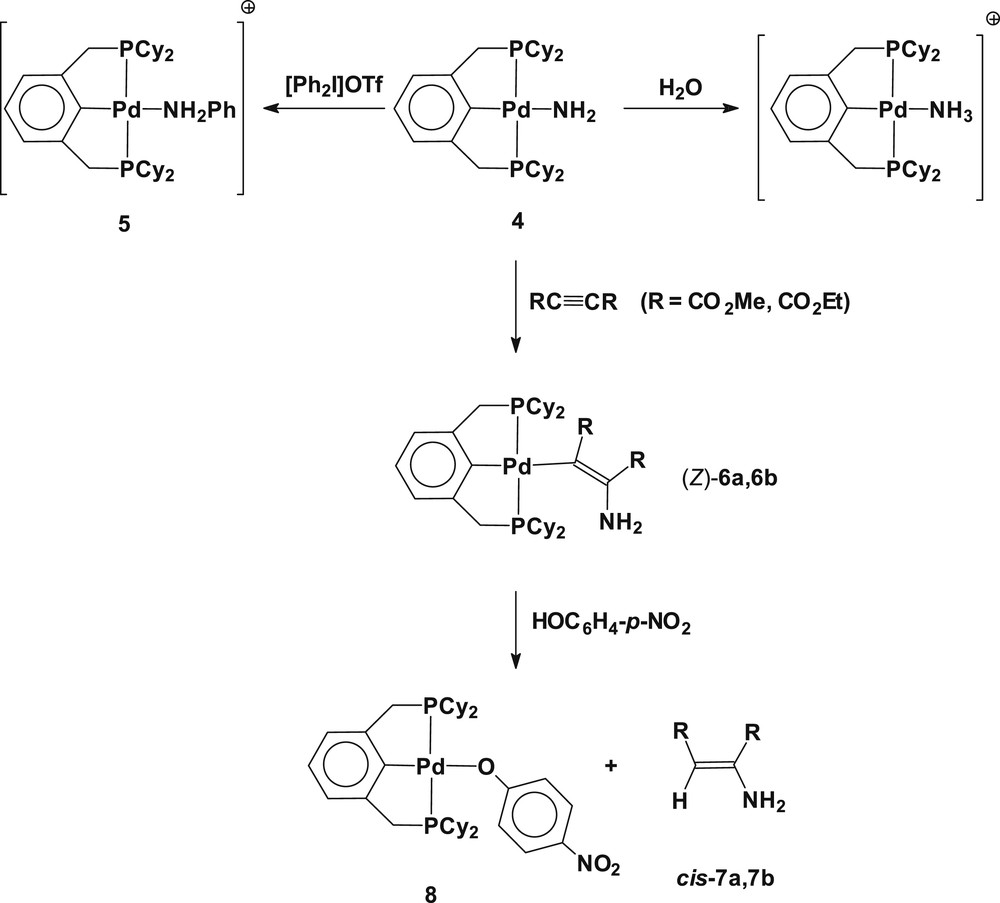
Reactions of (2,6-(Cy2PCH2)2C6H3)Pd(NH2) (4).
The monomeric amido complex 4 undergoes clean reactions with activated acetylene such as dialkyl acetylenedicarboxylate (RC≡CR; R = CO2Me, CO2Et) to produce the diastereospecific insertion derivative of Pd(2,6-(Cy2PCH2)2C6H3)(CR = CR(NH2)) (R = CO2Me (6a), CO2Et (6b)) (Scheme 3). Complexes 6a and 6b have been fully characterized by multinuclear NMR (1H, 13C, 31P) and FAB/MS spectroscopy. In solution, no coordination of the amino group in the complex has been observed, as evidenced by the 1H and 31P{1H} NMR spectroscopy; all signals were intact on addition of coordinating molecules such as pyridine and PPh3 to the d6-benzene solution of 6a and 6b. An attempt at isolation of complexes 6a and 6b was unsuccessful due to their high solubility in most organic solvents including n-pentane. Thus, removal of all volatiles from the solution under high vacuum resulted in yellow solids that afforded satisfactory FAB/MS spectroscopic data, displaying a parent molecular ion peak in good accordance with the calculated molecular weight in addition to the expected peaks due to molecular fragmentation. Although the absolute diastereomeric configuration of 6a and 6b could not be determined owing to failure of obtaining suitable crystals for an X-ray structural study, we assigned the stereochemistry of complexes 6a and 6b (Z)-isomer by performing subsequent reactions. A further reaction of 6a and 6b with an acidic phenol HOC6H4–p-NO2 produced only a single isomeric product cis-CHR = CR(NH2) (R = CO2Me (7a), CO2Et (7b)) with retention of configuration, along with the palladium(II) p-nitrophenoxide Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4–p-NO2) (8) (Scheme 3) (see Experimental section). For a control, reacting a d6-benzene solution of dialkyl acetylenedicarboxylate with ammonia produces an isomeric mixture of cis- and trans-(CHR = CR(NH2)) (see Experimental section). The diastereoselective formation of (Z)–Pd(2,6-(Cy2PCH2)2C6H3)(CR = CR(NH2)) (6a, 6b) resulted from the reaction of Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (4) and RC≡CR implies that the insertion reaction presumably involves a concerted pathway, apparently ruling out a dissociative nucleophilic addition. In view of the importance of utilizing ammonia as a substrate for the production of nitrogen-containing compounds [6], this study on the reaction profile for syn-insertion of activated acetylene into the PdNH2 bond in the title complex is noteworthy. Rare examples of syn-insertion of alkynes and alkenes into the PdN bonds have mostly been found in arylamido complexes [13]. Prior study on reaction of trans-Pd(PMe3)2(C6H5)(NHPh) with DMAD described syn-insertion of the C≡C triple bond into the PdNHPh bond to yield trans-Pd(PMe3)2(C6H5)((MeO2C)C = C(CO2Me)(NHPh)) [13a]. Inter- and intra-molecular syn-migratory insertion of alkenes into the Pd(II)-arylamido bonds in Pd(o-CH2P(t-Bu)2C6H4)(THF)(NR2) [13b] and (dppf)(C6H4F-p)Pd(N(C6H4F-p))((CH2)3CH = CH2) [13c], respectively, have also been demonstrated.
The formation of Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4–p-NO2) (8) generated from reaction of the vinyl complex (6a, 6b) and HOC6H4–p-NO2 has been verified by its independent synthesis from reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) and Na(OC6H4–p-NO2) in THF. The observed reactivity for the palladium(II) vinyl complexes toward HOC6H4–p-NO2 in the ligand exchange reaction reveals that the σ-vinylic ligand is more basic than the σ-phenoxide in the complexes, liberating stereoselective olefin derivatives with retention of configuration. The reaction of complex 4 with an activated olefin such as acrylonitrile was attempted, without giving a clean insertion product, instead producing polymeric species of acrylonitrile which precipitates in solution. The isolated polymer shows its characteristic absorption peaks for the ν(CN) at 2245 and 2204 cm−1 in the IR spectrum. No reaction of 4 with unactivated olefins such as 1-hexene and cyclohexene was observed, being presumably ascribed to an insufficient nucleophilicity of the coordinated amide along with the rigidity of the sterically hindered pincer ligand.
3 Conclusion
We have prepared novel parent-amido complexes of palladium(II) with bridging and terminal NH2. The monomeric palladium(II) amide Pd(2,6-(Cy2PCH2)2C6H3)(NH2) undergoes nucleophilic addition with activated acetylene RC≡CR (R = CO2Me, CO2Et) to yield diastereospecific vinyl complex (Z)-Pd(2,6-(Cy2PCH2)2C6H3)(CR = CR(NH2)). The produced vinyl complexes react with HOC6H4–p-NO2 to give Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4–p-NO2) along with olefin derivatives cis-CHR = CR(NH2) with retention of the diastereomeric configuration. The title complexes along with their reaction profiles toward the C≡C triple bond may provide useful information relevant to catalytic functionalization of unsaturated hydrocarbons with ammonia. Further study on catalytic hydroamination of unsaturated hydrocarbons with ammonia in the presence of the title complexes is currently underway.
4 Experimental
4.1 General
All preparations of air-susceptible compounds were carried out on a standard Schlenk line or in a glovebox under argon. PdCl2 was supplied by Kojima Chemicals Co., Ltd., and used without purification. AgOTf, dppe, α,α′-dibromo-m-xylene, CDCl3 and C6D6 were purchased from Aldrich Chemical Company, and used as supplied. Dicyclohexylphosphine was supplied by Strem Chemicals Inc. All other reagents were from various commercial companies. [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)]OTf and trans-[Pd(PEt3)2(Ph)(NH3)]OTf were prepared according to the literature [14]. IR spectra were recorded on a Bruker (Tensor 37) FT-IR spectrometer, as a pressed KBr pellets. 1H, 13C{1H} and 31P{1H} NMR spectra were measured on a Varian Gemini-2000 spectrometer. GC/MS analyses were carried out using an HP 6890 gas chromatograph equipped with an HP 5973 MSD and an HP-Ultra 1 column. FAB/MS and Elemental analyses were performed at Korea Basic Science Institute in Seoul, Korea.
4.2 Synthesis and reactions
4.2.1 Synthesis of anti-[Pd(Ph)(PEt3)(μ-NH2)]2 (2)
In a glovebox under argon atmosphere, a mixture of trans-[Pd(PEt3)2(Ph)(NH3)]OTf (500 mg, 0.85 mmol) and NaNH2 (150 mg, 3.8 mmol) was stirred in THF (20 mL) for 6 h at ambient temperature. Color of the reaction suspension was slowly changed from light grey to deep grey during the course of the reaction. The resulting suspension was filtered under vacuum to give a pale yellow solution. Removal of all volatiles from the filtrate under high vacuum resulted in yellow residues that were extracted with n-pentane (4 × 10 mL) to give a pale yellow solution. The volume of solution was reduced to ca. 10 mL to slowly give a colorless precipitates that were filtered, washed with cold n-pentane, and dried in vacuo. Yield 176 mg (65%). IR (KBr): ν(NH) = 3238, 3367 cm−1 (w, br). 1H NMR (C6D6): δ −1.35 br (4H, NH2), δ 0.93 m (18H, CH3), δ 1.09 m (12H, CH2), δ 7.08 t (2H, p-CH (Ph), 3J(HH) = 7.3 Hz), δ 7.26 t (4H, m-CH (Ph), 3J(HH) = 7.3 Hz), δ 7.78 d (4H, o-CH (Ph), 3J(HH) = 7.3 Hz). 13C{1H} NMR (C6D6): δ 8.22 s (CH3), δ 15.61 d (CH2, 1J(CP) = 26.7 Hz), δ 122.3, δ 127.1, δ 137.6. 31P{1H} NMR (C6D6): δ 20.2 s. Anal. Calcd for C24H44N2P2Pd2: C, 45.4; H, 6.98; N 4.41. Found: C, 45.1; H, 6.84; N, 4.20.
4.2.2 Synthesis of Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (4)
At a similar reaction condition as for complex 2, a mixture of [Pd(2,6-(Cy2PCH2)2C6H3)(NH3)]OTf (500 mg, 0.65 mmol) and NaNH2 (100 mg, 2.6 mmol) was stirred in THF (30 mL). The color of the reaction mixture was slowly changed from light grey to red during the course of the reaction. After 4 h, the resulting mixture was filtered under vacuum to give a red solution. Removal of all volatiles from the filtrate under high vacuum resulted in deep-red residues that were extracted with n-hexane (4 × 5 mL) to give a pale yellow solution. Removal of solvent from the extracted solution under high vacuum gave spectroscopically pure compound of Pd(2,6-(Cy2PCH2)2C6H3)(NH2). Yield 270 mg (68%). Spectral data for Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (4): 1H NMR (C6D6): δ −0.20 br (2H, NH2), δ 1.0–2.4 m (44H, Cy), δ 3.10 vt (4H, CH2, |2J(PH) + 4J(PH)| = 8.4 Hz), δ 7.08 m (3H, CH(aryl)). 31P{1H} NMR (C6D6): δ 50.7 s.
4.2.3 Reaction of Pd(2,6-(Cy2PCH2)2C6H3)(NH2) (4) with dialkyl acetylenedicarboxylate (RC≡CR; R = CO2Me, CO2Et) to yield (Z)-Pd(2,6-(Cy2PCH2)2C6H3)(CR=CR(NH2)) (R = CO2Me (6a), CO2Et (6b))
To a d6-benzene (0.3 mL) solution of 4 (15 mg, 0.024 mmol) in a 5-mm screw-capped NMR tube (Wilmad, 528-TR) was added a slight excess of dialkyl acetylenedicarboxylate (RC≡CR; R = CO2Me, CO2Et: 0.032 mmol, 0.1 mL of a diluted d6-benzene solution that was prepared by the addition of DMAD (46 mg) or DEAD (54 mg) into 1.0 mL of d6-benzene). The insertion product Pd(2,6-(Cy2PCH2)2C6H3)(CR = CR(NH2)) was quantitatively formed from the reaction, which was monitored by NMR spectroscopy. The product can hardly be isolated from the solution because of the high solubility in most organic solvents. Thus, the removal of all volatiles from the solution under high vacuum resulted in yellow solids that afforded satisfactory FAB/MS spectroscopic data. For Pd(2,6-(Cy2PCH2)2C6H3)((MeO2C)C = C(CO2Me)(NH2)) (6a): 1H NMR (C6D6): δ 1.0–2.4 m (44H, Cy), δ 3.49 s (3H, CH3), δ 3.73 s (3H, CH3), δ 3.10 dt (Ha, CH2; 2J(HaHb) = 17.6 Hz, |2J(PH) + 4J(PH)| = 7.9 Hz), δ 3.31 dt (Hb, CH2; 2J(HaHb) = 17.6 Hz, |2J(PH) + 4J(PH)| = 7.9 Hz), δ 3.89 br (2H, NH2), δ 7.21 m (3H, CH (aryl)). 13C{1H} NMR (C6D6): δ 50.53, δ 51.45 s (CO2CH3), δ 162.08, δ 176.40 (CO2CH3). 31P{1H} NMR (C6D6): δ 51.7 s. FAB/MS: observed m/z 761.22. Calcd for C38H59NO4P2Pd, 761.30. For Pd(2,6-(Cy2PCH2)2C6H3)((EtO2C)C = C(CO2Et)(NH2)) (6b): 1H NMR (C6D6): δ 1.0–2.4 m (44H, Cy), δ 1.08 t (3H, CH3, 3J(HH) = 7.1 Hz), δ 1.34 t (3H, CH3, 3J(HH) = 7.1 Hz), δ 4.12 q (2H, CH2, 3J(HH) = 7.1 Hz), δ 4.31 q (2H, CH2, 3J(HH) = 7.1 Hz), δ 3.10 dt (Ha, CH2; 2J(HaHb) = 17.6 Hz,|2J(PH) + 4J(PH)| = 8.3 Hz), δ 3.30 dt (Hb, CH2; 2J(HaHb) = 17.6 Hz,|2J(PH) + 4J(PH)| = 8.3 Hz), δ 3.94 br (2H, NH2), δ 7.21 m (3H, CH(aryl)). 13C{1H} NMR (C6D6): δ 14.47, δ 15.15 (CO2CH2CH3), δ 58.98, δ 60.30 (CO2CH2CH3), δ 162.08, δ 176.09 (CO2CH2CH3). 31P{1H} NMR (C6D6): δ 51.9 s. FAB/MS: observed m/z 789. 25. Calcd for C40H63NO4P2Pd, 789. 33.
4.2.4 Reaction of (Z)-Pd(2,6-(Cy2PCH2)2C6H3)(CR = CR(NH2)) (R = CO2Me (6a), CO2Et (6b)) with HOC6H4–p-NO2 to yield cis-CHR = CR(NH2) (R = CO2Me (7a), CO2Et (7b)) and Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4–p-NO2) (8)
To a d6-benzene (0.3 mL) solution of 6a or 6b (ca. 15 mg) in a 5-mm screw-capped NMR tube was added HOC6H4–p-NO2 (5 mg). The reaction proceeded quantitatively to produce cis-CHR = CR(NH2) (R = CO2Me (7a), CO2Et (7b)) and Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4–p-NO2) (8). All products were analyzed by 1H and 31P NMR, and GC/mass spectroscopy. The p-nitrophenoxide complex Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4–p-NO2) (8) can be independently prepared through an equimolar reaction of Pd(2,6-(Cy2PCH2)2C6H3)(OTf) and NaOC6H4–p-NO2 in THF. For cis-((MeO2C)CH = C(CO2Me)(NH2)) (7a): 1H NMR (C6D6): δ 3.15 s (3H, CH3), δ 3.44 s (3H, CH3), δ 5.77 s (1H, CH). GC/MS: m/z = 159, 128, 100, 68, 59. Calcd for C6H9NO4, 159.05. For cis-((EtO2C)CH = C(CO2Et)(NH2)) (7b): 1H NMR (C6D6): δ 0.78 t (3H, CH3, 3J(HH) = 7.13 Hz), δ 1.02 t (3H, CH3, 3J(HH) = 7.13 Hz), δ 3.79 q (2H, CH2, 3J(HH) = 7.13 Hz), δ 4.07 q (2H, CH2, 3J(HH) = 7.13 Hz), δ 5.84 s (1H, CH). GC/MS: m/z = 187, 142, 114, 86, 68. Calcd for C8H13NO4, 187.08. For Pd(2,6-(Cy2PCH2)2C6H3)(OC6H4–p-NO2) (8): IR (KBr): ν(NO) = 1583, 1303 cm−1 (sh, s). 1H NMR (C6D6): δ 0.9–2.1 m (44H, Cy), δ 2.84 vt (4H, CH2, |2J(PH) + 4J(PH)| = 8.4 Hz), δ 6.88 d (2H, 3J(HH) = 9.2 Hz), δ 8.49 d (2H, 3J(HH) = 9.2 Hz). 31P{1H} NMR (C6D6): δ 50.7 s. Anal. Calcd for C38H55NO3P2Pd: C, 61.5; H, 7.47; N 1.89. Found: C, 61.1; H, 7.52; N, 1.58.
4.2.5 Reaction of 4 with [Ph2I](SO3CF3) or [Ph2I]Cl
To a d6-benzene (0.3 mL) solution of 4 (15 mg) in a 5-mm screw-capped NMR tube was added a slight excess amount of [Ph2I]SO3CF3. The reaction produced a cationic aniline complex [Pd(2,6-(Cy2PCH2)2C6H3)(NH2Ph)]OTf, which was confirmed by its independent preparation from the reaction of Pd(2,6-(Cy2PCH2)2C6H3)OTf and NH2Ph, showing the identical 31P NMR resonance at δ 52.1 in d6-benzene. An attempt to isolate the cationic aniline species was unsuccessful due to the dissociation of the coordinated aniline converting to Pd(2,6-(Cy2PCH2)2C6H3)(OTf) (31P{1H} NMR (C6D6): δ 54.5). In the meantime, reaction of 4 with [Ph2I]Cl produced Pd(2,6-(Cy2PCH2)2C6H3)Cl (31P{1H} NMR (C6D6): δ 52.3) along with the liberation of NH2Ph (GC/MS: m/z = 93, 66, 39).
4.2.6 Reaction of dialkyl acetylenedicarboxylate with ammonia
Anhydrous ammonia gas was bubbled into a d6-benzene (0.3 mL) solution of dialkyl acetylenedicarboxylate (DMAD, DEAD; ca. 20 mg, respectively) for ca. 30 s in a 5-mm screw-capped NMR tube. The reaction produced an isomeric mixture of cis- and trans-(CHR = CR(NH2)) in a ratio of trans/cis = ca. 1.3. For trans-((MeO2C)CH = C(CO2Me)(NH2)): 1H NMR (C6D6): δ 3.42 s (3H, CH3), δ 3.59 s (3H, CH3), δ 4.80 s (1H, CH). For trans-((EtO2C)CH = C(CO2Et)(NH2)): 1H NMR (C6D6): δ 0.75 t (3H, CH3, 3J(HH) = 7.13 Hz), δ 1.01 t (3H, CH3, 3J(HH) = 7.13 Hz), δ 3.37 q (2H, CH2), δ 4.08 q (2H, CH2), δ 4.90 s (1H, CH). For cis-((MeO2C)CH = C(CO2Me)(NH2)) and cis-((EtO2C)CH = C(CO2Et)(NH2)): refer to Experimental section, § 4.2.4.
Acknowledgements
This work was supported by the Dongguk University Research Fund of 2013. The authors are grateful to Prof. M.S. Lah for an X-ray diffraction study.