

1 Introduction
The reactive imine ethoxycarbonyl-N-(2,2,2-trichloroethylidene)amine (1) (also named anhydrochloral-urethane), first described by Feist and Ulrich et al. [1,2], has been used in the past by several groups as a versatile starting material for a number of transformations. For example, treatment of Cl3C–CHN–CO2Et (1) with Grignard reagents gives Cl3C–CHRNHCO2Et (R = Me, Et, Pr, Bu, PhCH2, CH2CHCH2), which after hydrolysis and decarboxylation leads to α-amino acids [3]. Hetero-Michael addition of pyrazolinones to Cl3CCHNCO2R is reported to yield novel 1,3,4-substituted 2-pyrazolin-5-ones [4]. The synthesis of 5,6,7-substituted 1,2,4-triazolo[1,5-a]pyrimidines was achieved by the treatment of triazolopyrimidinols with 1 [5]. Several papers deal also with the utilisation of this imine as a heterodienophile for [4 + 2] Diels-Alder cycloadditions [6–9].
Our laboratory investigated in the late 90s the potential of the title compound as a reagent for the synthesis of heterocycles via 1,3-dipolar cycloaddition [10–12]. In this context we have described and discussed the unexpected formation of 4,4-dichloro-1,1-diphenyl-2-azabuta-1,3-diene [Ph2CN–CHCCl2] (4a) by cycloaddition of diphenyldiazomethane across the imine function of 1 at 65 °C in toluene [12]. We have demonstrated that the first step of the process consists of a dipolar 1,3-cycloaddition of the diazo-compound across the imine, leading to the Δ3-1,2,4-triazoline 2a. Then, extrusion of dinitrogen gives the transient azomethine ylide 3a. Subsequent elimination of ethyl chloroformate produces the 2-azadiene 4a in 70% yield (Scheme 1). Investigating the electronic structure and chemical properties of 4a, we have found that it presents an interesting reactivity towards various nucleophiles, such as alkoxides, thiolates, and the sodium salt of pyrrole or the cyanide anion [13–17]. More recently, some of these compounds were reacted as S,N-chelators with transition metal derivatives and allowed us to access new organometallic products via oxidative addition and cyclometallation reaction [18,19].
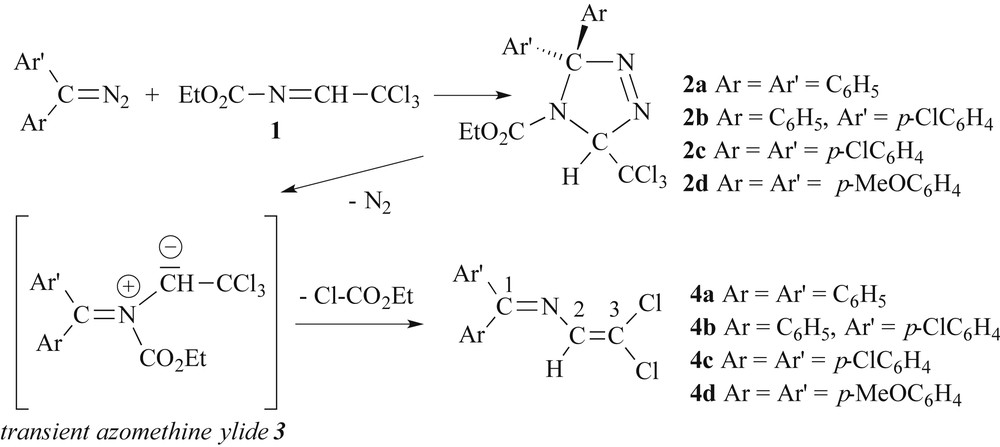
Reaction of diaryldiazomethanes with imine 1.
In order to modulate the electronic properties of this reactive 2-azadienic array, we now have investigated two different strategies to prepare functionalized derivatives of 4a to have a feedstock for further organic or organometallic transformations:
- (i) introduction of electron-pushing or withdrawing substituents at the para-positions of the phenyl rings.
- (ii) systematic investigation of the reactivity towards a wide series of thiolates SR− and amides NR2− to obtain functional ligands possessing both S,N or N,N donor sites for complexation studies.
We report herein the preparation of some new functionalized 1,1-diaryl-4,4-dichloro-2-azabutadienes and the rich reactivity of these π-conjugated compounds [20] towards various thiolate nucleophiles of the type NaSR. The molecular structures of some azadienic dithioethers resulting from the unpredictable attack of thiolates have been determined by means of X-ray diffraction studies. This investigation includes also a reactivity study of 4a toward the sodium salt of piperidine and the structural characterization of the resulting substitution product.
2 Results and discussion
2.1 Cycloaddition of diaryldiazomethanes Ar2CN2 across Cl3C–CHN–CO2Et
In analogy to the protocol using diphenyldiazomethane as a dipole, the cyloaddition of di(p-chlorophenyl)diazomethane, (p-chlorophenyl)phenyldiazomethane and di(p-methoxyphenyl)-diazomethane produces at 20 °C in toluene as solvent, the expected Δ3-1,2,4-triazolines 2b–d as colourless powders (see Scheme 1). Subsequent thermolysis of these heterocyclic compounds in toluene at 60° leads to (most probably via the transient azomethine ylides 3 [21]) the corresponding 1,1-diaryl-4,4-dichloro-2-azabutadienes 4b–d as pale-yellow stable solids. However, it is not necessary to isolate first the heterocyclic intermediates 2. If the imine 1 is treated with Ar2CN2 in warm toluene, in situ transformation affords straightforwardly the 2-azadienes 4. The crude residue of 4c is contaminated by 10–15% of tetrakis(p-chlorophenyl)diazine (p-ClC6H4)2CN–NC(p-ClC6H4)2 as a minor product, which can be separated by fractional crystallisation from hot ethanol [22].
As observed in the proton NMR spectrum of 4a, the vinylic hydrogen resonances of 4c and 4d are quite low-field shifted and appear as singlets at δ 7.07. Furthermore, two distinct singlets due to the methoxy groups are found at δ 3.83 and δ 3.87 in the spectrum of 4d. In the case of 4b, two singlets in a 2:1 ratio are detected at δ 7.03 and δ 7.01 in CDCl3 solution, respectively. Two singlets, attributed to the CN resonance, are also observed in the 13C{1H} spectrum at δ 167.0 and 166.9 in an approximate 2:1 ratio. Initially, we rationalized this finding by formation of two stereoisomers resulting from different approaches of the dissymmetric diaryldiazoalkane across the CN bond of imine 1 in the transition state. A careful examination under an optical microscope confirmed that the morphology of all crystals of 4b within this batch was quite homogenous, the melting point being sharp between 64–66 °C. Surprisingly, dissolving a single crystal in CDCl3 again gave rise to two sets of signals in the 2:1 ratio. A survey of the literature revealed that this phenomenon is most probably due to a hindered inversion at the imine nitrogen. For example, Jennings et al. investigated in detail a series of Schiff bases of type p-XC6H4(C6H5)CNR reminiscent to 4b, illustrated in Scheme 2 [23].

Invertomers resulting from N-inversion of the imine.
The activation barrier for this imine inversion has been estimated by means of dynamic NMR spectroscopy to be of some 30 kcal/mol, the value varying in function of substituent X. We suppose, that in the case of 4b, a similar inversion process may operate giving rise to a co-existence of E and Z invertomers in solution. Since this phenomenon is beyond the topic of this article, we did not undertake NMR studies at variable temperature.
2.2 Reactivity of 4a and 4c towards NaSAr, NaSC6H11, NaSMe and NaSnBu
Three strategies have been developed in the past to synthesise 2-azabuta-1,3-dienes with thioether substituents: (E)-1-methylthio-2-azabuta-1-3-diene-4-carbo-nitriles like A and B were prepared by the addition of thioamides to methoxymethylene compounds or ketene dithioacetals and subsequent methylation, [24] whilst 3-aza-2-(dimethylamino-4-(methylthio)-2,4-hexadiene (C) was obtained from treatment of N-(thiopropionyl)acetamidine with CH3I and subsequent deprotonation of the resulting N-ylidene amidinium salt [25]. The crystallographically characterized dithioether D and related compounds were obtained by alkylation of the dithiolates Na2[S2CC(R)–NCPh2] (R = CO2Me, CN) with CH3I or Br(CH2)nBr (n = 2, 3) [26].
In precedent papers we have demonstrated that 4a reacts under mild conditions in dry DMF with an excess of NaSR leading exclusively to the dithioether derivatives of type [Ph2CN–C(H)C(SR)2] (R = Ph, p-Tolyl, iPr), consistent with formal substitution of the two chloro-substituents on the vinylic C3 atom by SR−. Their crystal structures have been already published [14,15,19a]. An addition-elimination mechanism, initiated by nucleophilic addition to the C3 atom, elimination of chloride followed by addition of a second thiolate on the C3 atom and termination by dissociation of the remaining chloride has been suggested [14]. A mono-substituted thioether intermediate, [Ph2CN–C(H)C(Cl)(SPh)], in which the SR group is cis-situated relative to the imine nitrogen, has been structurally characterized [19b]. Note, that for vinylic substitutions also other mechanisms such as SRN1-type single-electron transfer may operate [27–29].
With the exception of 5a, 5b and 5d, which were isolated as viscous oils, all other derivatives (Scheme 3) formed yellow solids. An X-ray structure analysis has been carried out on a crystal of 5i grown from EtOH (Fig. 1). The s-trans conformation of the azabutadiene chain found in precursor 4a and in 5f, 5g and 5j is also observed in the solid-state structure of 5i. Metric parameters and crystallographic data are gathered in Tables 1 and 2. All in all, they resemble much to those of [Ph2CN–C(H)C(SPh)2] 5j [14] and [Ph2CN–C(H)C(p-STol)2] 5g [15] and deserve no further comment.
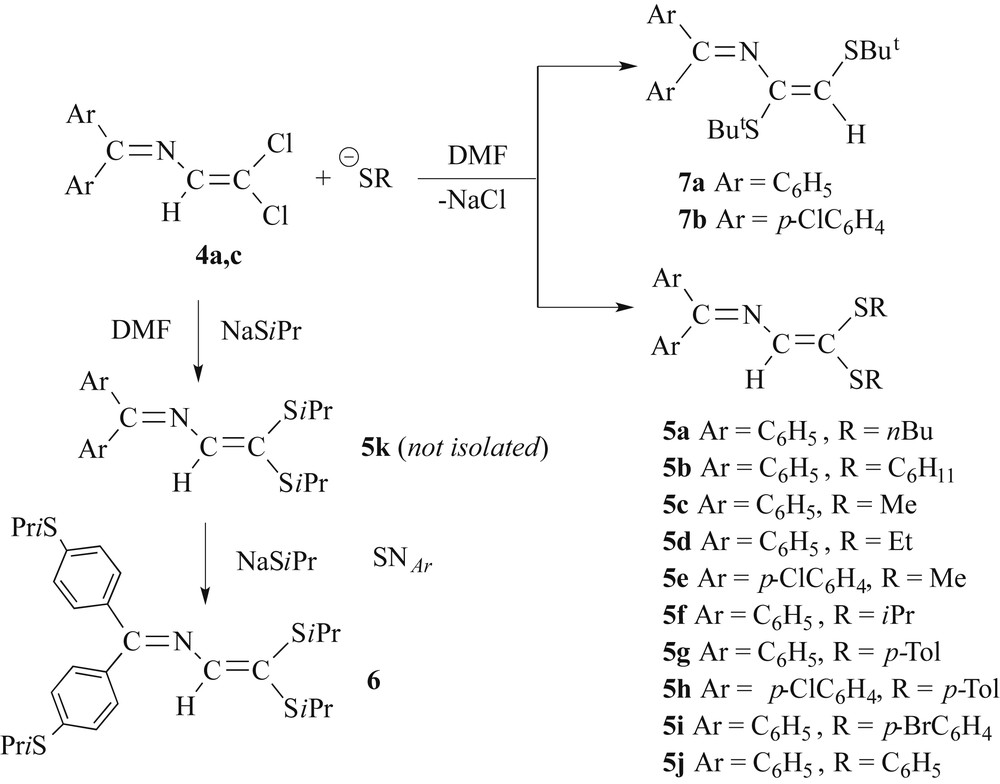
Reaction of 4 with aliphatic and aromatic thiolates.
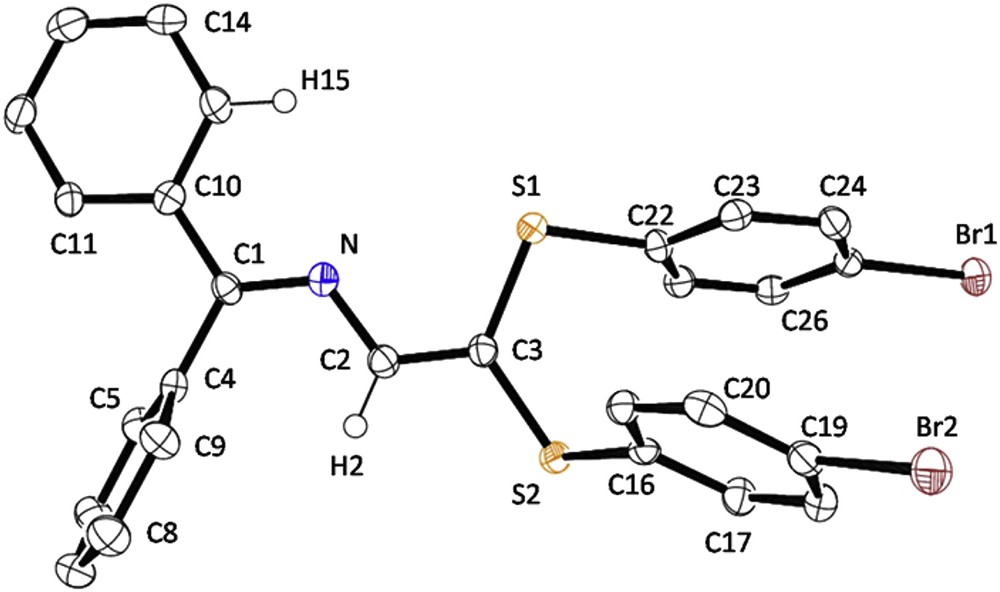
ORTEP view of 5i at the 50% probability level.
Significant metric parameters, distances (Å) and angles (°) for thiolate- and piperidine-substituted azabutadienes 5i, 7a,b, 10 as well as for the dinuclear copper complex 11.
| Compound | 5i 115K | 7a 293K | 7b 115K | 10 115K | 11 115K |
| C1–N | 1.304(4) | 1.274(3) | 1.286(2) | 1.298(1) | 1.302(6) |
| N–C2 | 1.379(5) | 1.411(3) | 1.424(2) | 1.385(1) | 1.396(6) |
| C2–C3 | 1.352(5) | 1.330(3) | 1.327(3) | 1.369(1) | 1.353(7) |
| C1–C(//)a | 1.469(5) | 1.487(4) | 1.484(2) | 1.483(1) | 1.490(7) |
| C1–C(⊥)a | 1.499(5) | 1.498(4) | 1.495(2) | 1.497(1) | 1.497(7) |
| C3–Xtransb | 1.749(4) | 1.769(3)c | 1.778(2)c | 1.407(1) | 1.778(5) |
| C3–Xcisb | 1.777(4) | 1.740(3) | 1.747(2) | 1.389(1) | 1.751(5) |
| N/C1/C(//)/C5 | −8.0 | 12.2 | 16.2 | −14.5 | 32.3(7) |
| N/C1/C(⊥)/C15 | 75.0 | 69.8 | 71.0 | −61.1 | 52.7(7) |
| N/C1/C(//)a | 117.4(3) | 118.0(3) | 118.6(2) | 117.85(10) | 119.3(4) |
| N/C1/C(⊥)a | 123.5(3) | 123.5(3) | 123.4(2) | 124.44(10) | 122.9(4) |
| C1/N/C2/C3 | −176.7 | −111.6 | −116.5 | 178.4(1) | 151.6(5) |
a C(//) and C(⊥) are the ipso carbon atoms of aryl groups, respectively, nearly coplanar and roughly perpendicular with respect to the plane of the azadiene chain.
b X = S or N, trans and cis refer to the N atom of the azabutadiene chain.
c C2–StBu.
Crystallographic and refinement data for compounds 5i, 7a, 7b, 10 and the Cu complex 11.
| Compound | 5i | 7a | 7b | 10 | 11 |
| Empirical formula | C27H19Br2NS2 | C23H29NS2 | C23H27Cl2NS2 | C25H31N3 | C54H74Br2Cu2N2S8 |
| Formula weight | 581.37 | 383.62 | 452.51 | 373.53 | 1294.06 |
| Temperature, K | 115(2) | 293(2) | 115(2) | 115(2) | 115(2) |
| Wavelength, Å | 0.71073 Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P | P21/a | P21/n | P | P |
| Unit cell dimensions (Å and °) | a = 6.8333(2) b = 12.6786(5) c = 14.1754(6) α = 102.577(1) β = 97.714(2) γ = 93.156(2) | a = 12.3662(5) b = 10.5268(4) c = 17.1480(8) α = 90 β = 97.109(2) γ = 90 | a = 9.3277(2) b = 14.8893(4) c = 17.3373(5) α = 90 β = 98.964(1) γ = 90 | a = 9.5449(4) b = 10.1818(4) c = 10.9957(3) α = 95.864(2) β = 96.571(2) γ = 103.203(1) | a = 10.4760(5) b = 11.9864(5) c = 12.9220(4) α = 101.519(2) β = 101.928(2) γ = 108.867(2) |
| Volume, Å3 | 1183.27(8) | 2215.11(16) | 2378.45(11) | 1024.39(7) | 1438.68(10) |
| Z | 2 | 4 | 4 | 2 | 1 |
| Density, calcd | 1.632 Mg/m3 | 1.150 Mg/m3 | 1.264 Mg/m3 | 1.211 Mg/m3 | 1.493 Mg/m3 |
| Linear Abs. Coef. Abs. correction | 3.618 mm−1 (Tmin 0.6707; Tmax 0.9312) | 0.247 mm−1 | 0.458 mm−1 | 0.071 mm−1 (Tmin 0.9824; Tmax 0.9915) | 2.455 mm−1 |
| F(000) | 580 | 824 | 952 | 404 | 667 |
| Crystal size (mm) | 0.12 × 0.07 × 0.02 | 0.15 × 0.15 × 0.05 | 0.25 × 0.2x0.1 | 0.25 × 0.15 × 0.12 | 0.12 × 0.02 × 0.02 |
| Theta range for data collection | 1.49 to 27.47° | 2.90 to 27.57° | 2.71 to 27.51° | 3.22 to 27.55 | 2.13 to 25.00° |
| Index ranges | −8 ≤ h ≤ 8 −16 ≤ k ≤ 16 −18 ≤ l ≤ 18 | −15 ≤ h ≤ 15 −13 ≤ k ≤ 12 −22 ≤ l ≤ 22 | −12 ≤ h ≤ 12 −19 ≤ k ≤ 17 −22 ≤ l ≤ 22 | −12 ≤ h ≤ 12 −13 ≤ k ≤ 13 −9 ≤ l ≤ 14 | −13 ≤ h ≤ 13 −15 ≤ k ≤ 15 −16 ≤ l ≤ 16 |
| Refl. collected | 16609 | 8505 | 9446 | 7680 | 38706 |
| Independent refl. (R(int)) | 5403 (0.0749) | 5029 (0.1045) | 5416 (0.0388) | 4585 (0.0165) | 6331 (0.1057) |
| Data/restraints/parameters | 5403/0/289 | 5029/0/235 | 3043/0/217 | 4585/0/253 | 6331/0/370 |
| Refl. gt [I>2σ(I)] | 4373 | 1639 | 3806 | 4068 | 4936 |
| GoF on F2 | 1.099 | 0.857 | 1.025 | 1.068 | 1.039 |
| Final R factors [I >2σ(I)] | R1 = 0.0452, wR2 = 0.0877 | R1 = 0.0530, wR2 = 0.0849 | R1 = 0.0412, wR2 = 0.0791 | R1 = 0.0427, wR2 = 0.1022 | R1 = 0.0426, wR2 = 0.1171 |
| R factors (all data) | R1 = 0.0626, wR2 = 0.0963 | R1 = 0.2351, wR2 = 0.1208 | R1 = 0.0736, wR2 = 0.0885 | R1 = 0.0500, wR2 = 0.1094 | R1 = 0.0524 wR2 = 0.1171 |
| Largest ρmax/ρmin (e.Å−3) | 0.482/−0.584 | 0.175/–0.216 | 0.296/−0.322 | 0.321/−0.176 | 1.589/−1.851 |
In contrast to the facile preparation of compounds [Ph2CN–C(H)C(SR)2] 5 bearing a Ph2CN imine function, the synthesis of derivatives [(p-ClC6H4)2CN–C(H)C(SR)2] 5e and 5k was less straightforward. [(p-ClC6H4)2CN–C(H)C(SMe)2] 5e could be isolated only in a quite modest yield of 20% as a yellow solid. Furthermore, chromatography of the crude residue of 5e provided a second fraction containing the tris(thioether) [(MeSC6H4)(p-ClC6H4)CN–C(H)C(SMe)2], albeit in a low yield of 3%. A high-resolution mass spectrum of this bright-yellow solid displayed a peak at m/z 379.66 (Fig. S1), whose simulation matches well with a composition of C18H18ClNS3, corroborating the formation of a triply SMe-substituted azabutadiene.
During the synthesis of the compound [(p-ClC6H4)2CN–C(H)C(SiPr)2] 5k, spectroscopic analysis did not allow to elucidate its structure unequivocally. Only in the case of [(p-ClC6H4)2CN–C(H)C(STol-p)2] 5h, a high-yield synthesis could be performed. Since aromatic halides are known to undergo nucleophilic substitutions of the SNAr type with highly nucleophilic aliphatic thiolates NaSMe and NaSiPr, we reacted 4c with a 10-fold excess of NaSiPr in DMF under gentle heating. Indeed, after workup, we succeeded in isolating the azadienic tetrakisthioether [(iPrSC6H4)2CN–C(H)C(SiPr)2] 6 with 48% yield in the form of a yellow solid (Scheme 3) [30,31].
2.3 Reactivity of 4a and 4c towards sodium tert-butylthiolate
We extended the reactivity study of 4a,c towards thiolates also on the reaction with NaStBu in DMF as solvent. Surprisingly, the spectroscopic features of the substitution products 7, which were isolated after workup as orange-red stable crystalline solids, diverged much from those of 5. For example, the vinylic H resonance of 7a and 7b were detected at δ 5.98 and 6.01 ppm in the 1H NMR spectrum, whereas the vinylic H atom of 5 gives rise to a singlet at a much lower field of about 7.1 ppm. The tBu groups appear as a set of close, but distinct singlets for 7a at δ 1.39 and 1.40 ppm (δ 1.35 and 1.43 for 7b). In the case of 7a, comparison of the experimental chemical shift (5.98 ppm) with the values of the chemical shifts at 5.94 (trans or E with respect to both tBu substituents) and 6.61 (cis or Z) ppm calculated by the incremental method of Pascual, [32] allowed us to assign E-stereochemistry around the olefinic double bond. This E-stereochemistry was furthermore ascertained by X-ray crystallography.
X-ray quality crystals of 7a,b were grown from EtOH and the molecular structures are depicted in Fig. 2. The outstanding information got from concerns the position of the StBu substituents. One StBu is found, as in all our thioether compounds 5, at the C3 atom and in the cis-position relative to the imine nitrogen. However, and for the first time in this series, we observe the second SR substituent on the C2 atom. The torsion C1–N–C2–C3 angles are far from 180° (7a 111.6(2)°, 7b 116.5(2)°, Table 2), approaching a limit of 90° for which both formal double bonds are perpendicular and their conjugation is almost completely broken. This is nicely demonstrated by the N–C2 bonds (7a 1.411(3) Å, 7b 1.424(2) Å), which are longer than those observed in the s-trans structures where the C2 atom bears the hydrogen atom (see other compounds in Table 1). Thus the s-trans conformation of the azabutadiene chain seen for 5 is strongly perturbed. Such a large distortion from ideal s-trans conformation has been already observed in other 2-aza-1,3-butadiene structures substituted at the C2 atom [33–40] The dihedral angles between “nearly parallel” phenyl ring C4–C9 and the three atom C1–N–C2 plane of the azadienic chain are equal to 15.0(3)° (7a) and 18.6(2)° (7b) with the corresponding N/C1/C4/C5 torsion angles of 12.2° and 16.2°, respectively. These experimental values fall in the range observed for compound 5i. The N/C1/C4 and N/C1/C10 angles in the imine part of the molecules also have values close to those found in 5.

ORTEP views of 7a (left, 20% probability level) and 7b (right, 50% probability level).
Of course the question arises, why 4 exhibits a different substitution pattern in the case of NaStBu compared to the other sodium thiolates, including even steric demanding thiolates such NaSiPr and NaSC6H11. At the first glance, one may suspect a strong sterical repulsion between two geminal -StBu groups residing on the terminal carbon atom of a hypothetical isomer [Ar2CN–C(H)C(StBu)2](7′). However, a literature survey reveals that polythioethers with two gem-StBu groups attached on the same carbon atoms exist, [41] for instance the allene [(tBuS)2CCC(Ph)(StBu)] [42]. Another example is the structurally characterized butatriene-type tetrakisthioether [(tBuS)2CCCC(StBu)2] [43]. But in all these examples, the adjacent carbon atom is an allenic one of the C type. But there is also a report on the diene 7,8,8-tris[(tert-butyl)thio]-6-vinylpentafulvene [(tBuS)2CC(tBuS)–C(H)C5H4] [44].
Let's have a look on the simplest model compound, e.g. dichlorovinylidene. Truce et al. have studied the reactivity of Cl2CCH2 toward NaSTol-p and NaStBu [45,46]. In the case of the more nucleophilic NaStBu reagent, they established an addition-elimination pathway via a transient alkyne species rationalizing the formation of cis-1,2-bis(tert-butylmercapto)ethene as the sole product [45,46]. Some intermediates such as 1,1-dichloro-2-(tert-butylmercapto)ethane and trans-1-chloro-2-(tert-butylmercapto)ethene could be isolated. Later work of Tanimoto et al. confirmed that the reaction of dichlorovinylidene with sodium phenylthiolate in DMF affords exclusively cis-1,2-bis(phenylmercapto)ethane [47–49]. We propose a similar addition-elimination mechanism explaining the formation of 7 (Scheme 4).
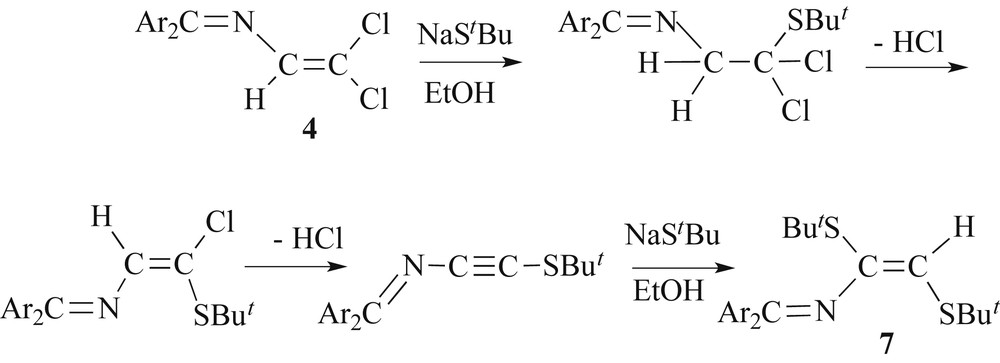
Possible mechanism of the conversion of 4 to 7 in the presence of NaStBu.
More recently our group demonstrated that upon the reaction of dibromovinylferrocene [Br2CC(H)-Fc] with NaStBu and NaSAr (Ar = Ph, p-tol) the structurally characterized rearrangement products Z-[(tBuS)(H)CC(H)-Fc] and Z-[(ArS)(H)CC(SAr)-Fc] were formed [50a]. In a similar manner, treatment of dibromovinyl[2.2]paracyclophane [Br2CC(H)-PCP] with excess of NaSAr in DMF gives rise to the vicinal dithioethers Z-[(ArS)(H)CC(SAr)-PCP] [50b]. Therefore one may conclude from the above discussion that the formation of the azabutadienes 5 bearing two geminal SR functions could be considered as an exceptional case, whereas the rearrangement encountered in the case of NaStBu leading to 7 corresponds rather to the “classical” pathway. We suppose that a combination of both steric and electronic factors is decisive for the formation of 7 using the bulky and highly nucleophilic NaStBu. The NaSnBu reagent leading to 5a should have a comparable basicity and nucleophilicity with NaStBu, but is doubtlessly sterically less demanding.
2.4 Reactivity of 4a towards sodium ethylthiolate
In analogy to the synthesis of compounds 5, the reaction of 4a with NaSEt in DMF at room temperature afforded exclusively [Ph2CN–C(H)C(SEt)2] 5d, as confirmed by the presence of the vinylic hydrogen as a singlet at δ 7.10 ppm and two sets of SEt groups in the 1H NMR spectrum. However, when the reaction of 4a (Scheme 5) was conducted with an excess of NaSEt under reflux in neat ethanethiol for 4 days, imine [Ph2CN–C(H)(SEt)–CCl2H] 8 was isolated as a yellow solid. The constitution of 8 has been elucidated from mass spectroscopy and NMR data. Both the 1H (Fig. S2) and 13C NMR spectra of 8 reveal the presence of only one thioethyl group. This unexpected product results probably from formal EtSH addition across the CC bond of 4a, similar to the first step of the reaction sequence shown in Scheme 4.
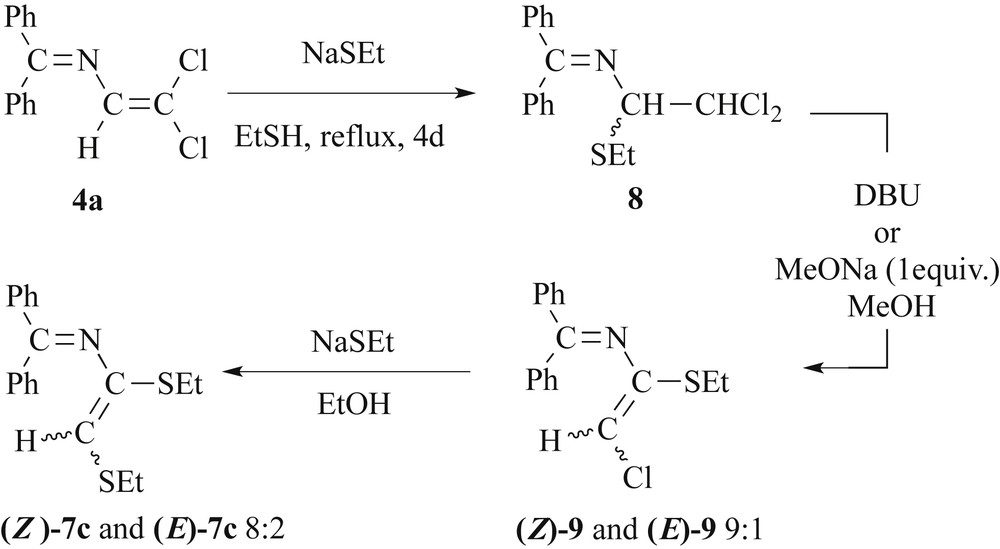
Reactivity of 4a towards sodium ethylthiolate.
It seems that the basicity of NaSEt does not permit the elimination of HCl. However, the reaction of compound 8 in toluene with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or an equivalent of NaOMe in methanol yields quantitatively the dehydrochlorinated compound 9. We identified a mixture of two diastereomers (Z)-9/(E)-9 in a 9:1 ratio. The proton NMR spectrum allows distinguishing two vinylic protons at 5.22 and 5.52 ppm. By comparison with the chemical shifts at 5.23 and 5.51 ppm respectively, calculated by the incremental method of Pascual, [32] we can assign the major compound (singlet at 5.52 ppm) to the stereoisomer (Z)-9. The synthesis of the azabutadiene 7c, which is structurally related to 7a,b, was achieved by reacting 9 with an excess of NaSEt in ethanol. The proton NMR spectrum revealed again the presence of a mixture of two stereoisomers (Z)-7/(E)-7 in an approximate 8:2 ratio. Two vinylic protons are observed at 5.20 and 5.47 ppm. Again, the incremental method of Pascual allowed a stereochemical assignment of the resonances. For the dominating stereoisomer (Z)-7c, a chemical shift of 5.23 ppm was calculated. For the vinylic hydrogen of the minor isomer (E)-7c, a chemical shift of 5.47 ppm was calculated, demonstrating the excellent match with the experimental data.
2.5 Reactivity of 4a towards the sodium salt of piperidine
With the aim to prepare amine-substituted azabutadienes related to 5 and 7, we have reacted 4a with secondary amides X2N−Na+ (X = Ph, Et) as well as with NaNH2. But despite varying the reaction conditions, we failed to isolate any product. Fortunately, treatment of 4a with cyclic amides was more successful. We have communicated that the reaction of 4a with the sodium salt of pyrrole leads, through an addition-elimination process, to (E)-4-chloro-1,1-diphenyl-3-(1-pyrrolyl)-2-azabuta-1,3-diene, in which the chloro and the N-pyrrole substituents are in a trans-configuration [16]. For comparison, we treated 4a with the in situ prepared sodium salt of piperidine using neat piperidine as the reaction medium. After work-up, a yellow solid [Ph2CN–C(H)C(NC5H10)2] (10) was isolated in 45% yield (Scheme 6).
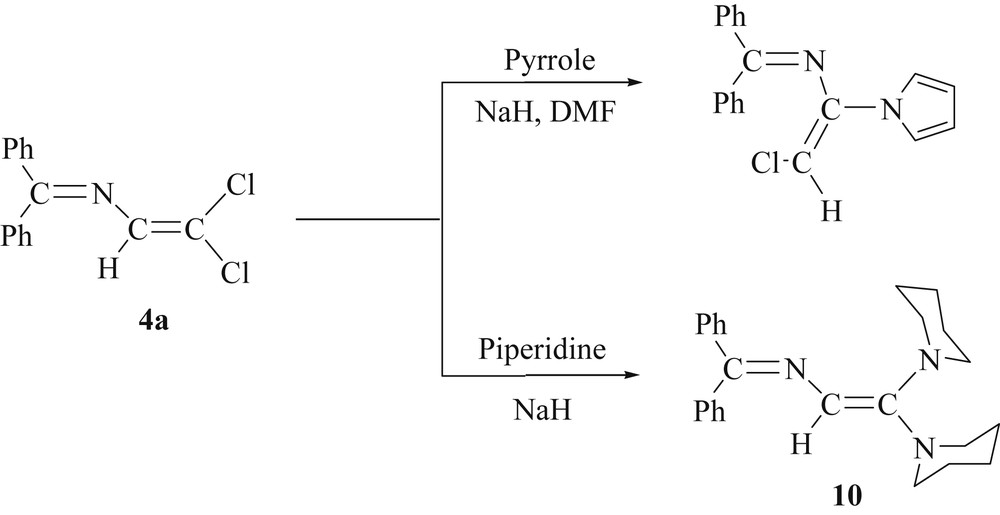
Reaction of 4a with cyclic amides.
Since the spectroscopic data, notably the chemical shift of the vinylic H-proton resonating at δ 5.40 ppm did not allow an unambiguous assignment of the substitution pattern of the azadienic chain, we have grown single-crystals of 10 from ethanol and determined the molecular structure of this piperidino-functionalized 2-azabutadiene. An ORTEP plot depicted in Fig. 3 confirms that the molecular geometry and substitution pattern correspond to that of [Ph2CN–C(H)C(OPh)2] [14] and [Ar2CN–C(H)C(SR)2] (5). Again, one aryl group is almost coplanar with the transoid azabutadiene array (N1/C1/C4/C5 torsion angle −14.5(2)°, Table 1). The torsion angle of the azadiene array (C1/N1/C2/C3) reaches 178.4(1)°. Both piperidine substituents on the C3 atom adopt a chair conformation. The mean C–N distance of the CC(NC5H10)2 moiety bearing the two bulky piperidyl groups amounts to 1.395(2) Å and compares well with that reported for methyl-3,3-dipiperidinodithioacrylate [MeSC(S)–C(H)C(NC5H10)2] (1.35(4) Å) [51] as well as with other structures reported in this article.
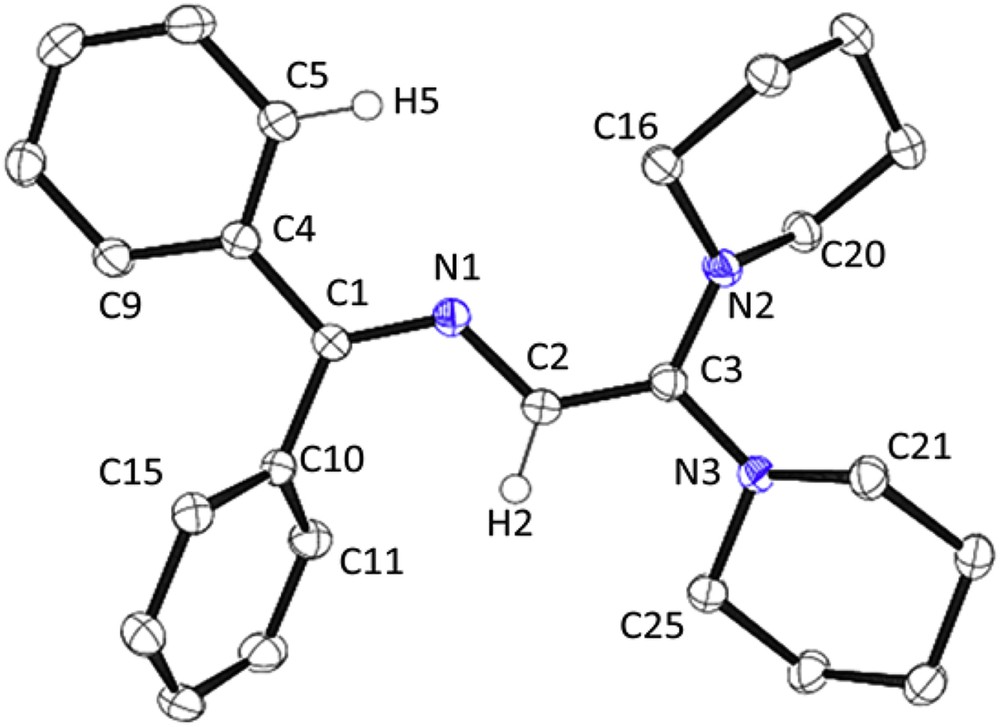
ORTEP view of 10 at the 50% probability level. Hydrogen atoms (except H2 and H5) are omitted for clarity.
2.6 Coordination of 6 on CuBr yielding a 16-membered macrocyclic complex
As stated in the introduction, one of the aims of this work was the preparation of a wide range of potentially bi- or poly-dentate unsaturated ligands for coordination chemistry. In a preliminary study on the coordination properties of tetrathioether 6, we reacted this ligand with CuBr in a 1:1 metal-to-ligand ratio. A single-crystal diffraction study (Fig. 4) indicates that from four available sulphur coordination sites in the ligand, only one aryl-bound SiPr group and one vinylic SiPr group are involved in the complexation of 6 on CuBr leading to the original dinuclear 16-membered macrocyclic complex [{Cu2Br2{p-iPrSC6H4)2CN–C(H)C(SiPr)2}2] (11). The cycle is built over the crystallographic symmetry centre in the P space group. Consequently, the central phenyl rings (C4 through C9 between the Cu/Cu* atoms) are parallel but slipped with respect to each other. The distance between these two planes is close to 3.4 Å and may so suggest a kind of intra-molecular π stacking (Fig. S3) as encountered in cyclophanes.

ORTEP view of the dinuclear macrocyclic complex 11 at the 30% probability level. Selected bond lengths [Å] and angles [°]: Cu–Br 2.3635(8), Cu–N 2.120(4), Cu–S1 2.3537(13), Cu–S3 2.3382(14), N–Cu–S1 83.88(11), N–Cu1–S3 100.28(11), N–Cu–Br 123.81(11), S1–Cu–S3 103.18(5), S1–Cu–Br 118.38(4), S3–Cu–Br 120.19(4), see also Table 1 for the parameters inside the ligand.
3 Conclusion and perspectives
We have shown that p-aryl-substituted 2-azadienes of type [Ar2CN–C(H)CCl2] (4) (Ar = Ph, p-C6H4Cl, p-C6H4OMe) are accessible by 1,3-dipolar cycloaddition of Ar2CN2 across Cl3C–CHN–CO2Et and observed that the nucleophilic attack of RS− and NaNC5H10 leads preferentially to [Ar2CN–C(H)C(SR)2] (5) and [Ph2CN–C(H)C(Pip)2] (8) bearing the SR and NR2 substituents at the C3 carbon. However, the fact that the nucleophilic attack of NatSBu affords the dithioethers [Ar2CN–C(StBu)C(H)(StBu)] (7a,b) as sole products demonstrates that the outcome of the reaction is difficult to predict and depends in a sensitive manner on several parameters. This was illustrated in the reaction of 4a with EtSNa in DMF or EtSH as solvents, leading to the formation of mono- and di-substituted compounds 5d or 8, respectively. The organic chemistry of the intermediate triazolines heterocycles is currently extended to the preparation of mixed alkyl-aryl systems of the type [Ar(Alkyl)CN–C(H)CCl2].
The straightforward preparation of ligands 5 and 7 permits furthermore a promising development in coordination chemistry, where a subtle fine-tuning of the stereoelectronic parameters exerted by the substituents −SR is of great importance. The design of the polydentate tetrakisthioether 6 and its coordination on CuBr giving rise to the unusual macrocyclic compound 11 demonstrates the potential of this ligand for the assembly of polymetallic systems and metal-organic frameworks [52]. In this context, we are currently extending the reactivity studies of 4 towards other nucleophiles and are exploring the coordination chemistry vis-à-vis numerous transition metals like Re, Mn, Cu, Ag, Ru and Fe and are investigating the electrochemical and photophysical properties of resulting mono- and di-nuclear systems. The potential of these polydentate ligands to act as C,N,S and C,N,N pincer ligands after cyclometallation reactions with Pd(II), Pt(II) and Ir(I), the electronic and molecular structures of the resulting organometallic compounds possessing a reactive covalent metal-aryl bond towards insertion reactions will be the topic of a forthcoming paper.
4 Experimental
4.1 General methods
The NMR spectra were recorded on a Bruker Avance 300 apparatus (300.13 and 75.1 MHz for 1H and 13C) in CDCl3, unless otherwise stated. FT-IR analysis was performed on a Shimadzu IR Affinity-1 apparatus (KBr, ν in cm−1). Melting points were obtained on a Büchi (SMP10) capillary melting point apparatus. UV–vis spectra were measured with a VARIAN-Cary 100 spectrophotometer in CH2Cl2 at room temperature, λmax in nm (ɛ in M−1 cm−1). The hydrazones Ar2CN–NH2 were prepared according to the literature and converted to Ar2CN2 using yellow HgO [53–55]. The thiols were purchased commercially from Aldrich and Alfa Aesar.
4.2 General procedure for the synthesis of the Δ3-1,2,4-triazolines
An equimolar mixture (10 mmol) of the diaryldiazomethanes and N-ethoxycarbonyl-N-(2,2,2-trichloroethylidene)amine in 10 mL of toluene or dichloromethane, was stirred at room temperature for 2–3 days. The solvent was then evaporated, and the residue was filtered and washed with ethanol.
4.2.1 5-Trichloromethyl-3-p-chlorophenyl-4-ethoxycarboxylate-3-phenyl-Δ3-1,2,4-triazoline 2b
Colourless powder; Yield: 68%; mp 78 °C; 1H NMR: δ = 0.68 (s br, 3H, CO2CH2CH3), 3.75 (s br, 2H, CO2CH2CH3), 6.75–7.72 (m, 10H, 9 Ar–H + 1CH); 13C NMR: δ = 14.2 (q, J = 127.5 Hz, CO2CH2CH3), 62.9 (t, J = 148.5 Hz, CO2CH2CH3), 96.0 (s, CCl3), 109.6 (d, J = 163.0 Hz, C-5), 122.5 (s, C-3), 125.0–140.0 (6 s, 9 CHar), 135.7–137.0 (2s, 2 Car), 155.0 (s, CO); IR: 1710 (CO); C18H15Cl4N3O2 (447.14): Anal. Calcd C 48.35, H 3.38, N 9.40, Cl 13.72; found: C 48.23, H 3.41, N 9.34, Cl 13.67.
4.2.2 5-Trichloromethyl-3,3-bis(4-chlorophenyl)-4-ethoxycarbonyl-Δ3-1,2,4-triazoline 2c
Colourless powder; Yield: 60%; mp 119 °C; 1H NMR (DMSO-d6): δ = 0.80 (s br, 3H, CO2CH2CH3), 3.85 (s br, 2H, CO2CH2CH3), 7.00–7.90 (m, 8H, Ar–H), 8.02 (s, 1H, CH); 13C NMR (DMSO-d6): δ = 13.2 (q, J = 127.0 Hz, CO2CH2CH3), 62.7 (t, J = 148.0 Hz, CO2CH2CH3), 96.4 (s, CCl3), 109.0 (d, J = 164.0 Hz, C-5), 113.8 (s, C-3), 125.0–132.0 (4 s, 4 CHar), 131.6–136.1 (4 s, 4 Car), 153.5 (s, CO); IR: 1706 (CO); C18H14Cl5N3O2 (481.60): Anal. Calcd C 44.89, H 2.93, N 8.73, Cl 36.81; found: C 45.02, H 2.98, N 8.48, Cl 36.96.
4.2.3 5-Trichloromethyl-4-ethoxycarbonyl-3,3-bis(4-methoxyphenyl)-Δ3-1,2,4-triazoline 2d
Pale yellow powder; Yield: 75%; mp 99 °C; 1H NMR: δ = 0.75 (s br, 3 H, CO2CH2CH3), 3.75 (s br, 2 H, CO2CH2CH3), 3.77 (s, 3 H, OCH3), 3.85 (s, 3 H, OCH3), 6.50–7.80 (4 d, 8H, Ar–H), 7.35 (s, 1 H, CH); 13C NMR: δ = 13.5 (q, J = 126.3 Hz, CO2CH2CH3), 55.0 (q, J = 144.0 Hz, 2C, OCH3), 62.5 (t, J = 148.0 Hz, CO2CH2CH3), 96.3 (s, C-3), 113.2 (d, J = 151.0 Hz, C-5), 126.0–132.0 (5 s, 4 CHar + Car), 133.7 (s, Car), 155.4 (s, Car–O), 156.0 (s, Car–O), 159.5 (s, CO); IR: 1702 (CO); C20H20Cl3N3O4 (472.76): Anal. Calcd C 50.76, H 4.23, N 8.88, Cl 22.52; found: C 50.92, H 4.36, N 5.56, Cl 22.73.
4.3 General procedure for the synthesis of azadienes 4
An equimolar mixture (10 mmol) of the diazo compound and of N-ethoxycarbonyl-N-(2,2,2-trichloroethylidene)amine in 10 mL of toluene is stirred at 65 °C for 3 days. Then, the toluene is evaporated and the oily residue is chromatographed on silica or on neutral alumina, using a CH2Cl2-petroleum ether mixture (50:50) as the eluant. Recrystallization from ethanol provided 4 in the form of pale yellow crystals.
4.3.1 4,4-Dichloro-1-(4-chlorophenyl)-1-phenyl-2-azabuta-1,3-diene 4b
Yellow crystals; Yield: 52%; mp 65 °C; IR: 1545 (CN), 1600 (CC); UV–vis: 256 sh (11100), 307 (18250); C15H10Cl3N (310.61): Anal. Calcd C 58.00, H 3.25, Cl 34.24, N 4.51; found: C 57.87, H 3.29, Cl 34.11, N 4.53.
Major isomer: 1H NMR: δ = 7.03 (s, 1H, CH), 7.10–7.80 (m, 10H, Ar–H + CH) ppm; 13C{1H} NMR: δ = 124.0 (C, C-4), 127.5–137.1 (9C, Car), 137.3 (C, C-3), 167.0 (C, CN) ppm.
Minor isomer: 1H NMR: δ = 7.02 (s, 1H, CHb), 7.10–7.80 (m, 10H, Ar–H + CHa); 13C{1H} NMR: δ = 124.0 (C, C-4), 127.5–137.1 (9C, CHar), 138.2 (C, C-3), 166.9 (C, CN).
4.3.2 4,4-Dichloro-1,1-bis(4-chlorophenyl)-2-azabuta-1,3-diene 4c
Yellow needles; Yield: 48%; mp 119 °C; 1H NMR (acetone-d6): δ = 7.07 (s, 1H, CH), 7.27–7.80 (4d, 8H, Ar–H); 13C NMR (acetone-d6): δ = 124.7 (s, C-4), 128.0–131.0 (4 s, 4CHar), 133.2–137.5 (4s, 4Car), 134.7 (d, J = 172.0 Hz, C-3), 165.5 (s, CN); IR: 1580 (CN), 1660 (CC); UV–vis: 270 (13900), 316 (22600); C15H9Cl4N (345.05): Anal. Calcd C 52.21, H 2.63, Cl 41.10, N 4.06; found: C 52.05, H 2.63, Cl 41.23, N 3.98.
4.3.3 4,4-Dichloro-1,1-bis(4-methoxyphenyl)-2-azabuta-1,3-diene 4d
Yellow powder; Yield: 58%; mp 117 °C; 1H NMR: δ = 3.83 (s, 3 H, OCH3), 3.87 (s, 3 H, OCH3), 6.80–7.80 (4d, 8H, Ar–H), 7.07 (s, 1 H, CH) ppm; 13C NMR: δ = 55.2 (q, J = 144.0 Hz, OCH3), 55.3 (q, J = 144.0 Hz, OCH3), 113.0–136.0 (5 s, 4 CHar + C-3), 121.3 (s, C-4), 127.7–131.8 (2s, 2 Car), 160.2–162.0 (2s, 2 OCar), 167.9 (s, CN). IR: 1603 (CN), 1634 (CC); UV–vis: 293 (17400); C17H15Cl2NO2 (336.23): Anal. Calcd C 60.73, H 4.50, Cl 21.11, N 4.17; found: C 60.59, H 4.55, Cl 21.47, N 4.20.
4.4 General procedure for the synthesis of 2-azabuta-1,3-dienes 5, 6, and 7.
1,1-Diaryl-4,4-dichloro-2-azabuta-1,3-diene (1.1 mmol) was stirred with an excess of thiolate (10 mmol) in dry DMF (10 mL). The reaction mixture was stored at room temperature for 8 h, then poured into water (100 mL) and extracted with diethyl ether (150 mL). The organic solution was washed three times with water, dried over anhydrous sodium sulfate and evaporated. The crude residue was recrystallized from ethanol or hexane.
4.4.1 4,4-Bis(butylthio)-1,1-diphenyl-2-azabuta-1,3-diene 5a
Yellow oil; yield: 76%; 1H NMR: δ = 0.75 (t, J = 7.3 Hz, 3H, –CH3), 0.82 (t, J = 7.3 Hz, 3H, –CH3), 1.16–1.26 (sex, J = 7.5 Hz, 2H, –CH2–CH3), 1.30–1.46 (m, 4H, –CH2–), 1.51–1.61 (quin, J = 7.3 Hz, 2H, –CH2–C2H5), 2.55 (t, J = 7.3 Hz, 2H, –SCH2), 2.93 (t, J = 7.3 Hz, 2H, –SCH2), 7.00 (s, 1H, CH), 7.06–7.64 (m, 10H, Ar–H); 13C{1H} NMR: δ = 13.7 (CH3), 13.9 (CH3), 21.8–22.0 (2C, CH2CH3), 31.3–32.2 (2C, CH2C2H5), 32.9–33.1 (2C, SCH2), 128.2–139.4 (9Car); 139.5 (C, CH), 164.4 (C, CN); IR: 1599 (CN), 1663 (CC); UV–vis: 255 (15100), 376 (13700); C23H29NS2 (383.61): Anal. Calcd C 72.01, H 7.62, N 3.65, S 16.72; found: C 71.98, H 7.83, N 3.67, S 16.70.
4.4.2 4,4-Bis(cyclohexylthio)-1,1-diphenyl-2-azabuta-1,3-diene 5b
Yellow oil; yield: 80%; 1H NMR: δ = 1.25–2.34 (m, 20H, CH2 of cyclohexyl), 2.90–3.04 (m, 2H, S–CH), 7.13–7.84 (m, 11H, Ar–H + CCH); 13C{1H} NMR: δ = 25.3–26.0 (6C, CH2 of cyclohexyl), 32.7 (2C, S–CH–CH2), 33.4 (2C, S–CH–CH2), 45.1 (C, S–CH); 45.9 (C, S–CH); 127.3–130.2 (12Car + CCH); 141.3 (CH), 164.8 (C, CN); UV–vis: 256 (12900), 378 (11200). ; C27H33NS2 (435.69): Anal. Calcd C 74.43, H 7.63, N 3.21, S 14.72; found: C 73.56, H 7.49, N 3.03, S 14.56.
4.4.3 4,4-Bis(methylthio)-1,1-diphenyl-2-azabuta-1,3-diene 5c
Yellow powder; Yield: 68%; mp 76 °C; 1H NMR: δ = 2.25 (s, 3H, SCH3), 2.55 (s, 3H, SCH3), 7.04 (s, 1H; CH), 7.22–7.79 (m, 10H; Ar–H); 13C{1H} NMR: δ = 16.3 (C, SCH3), 17.7 (C, SCH3), 128.1–137.3 (8Car + CCH), 139.5 (C, CH), 163.7 (C, CN); IR: 1554 (CN), 1589 (CC); UV–vis: 248 (9200), 366 (9700); C17H17NS2 (299.08): Anal. Calcd C 68.18, H 5.72, N 4.68, S 21.42; found: C 68.43, H 5.70, N 4.44, S 21.31.
4.4.4 4,4-Bis(ethylthio)-1,1-diphenyl-2-azabuta-1,3-diene 5d
Yellow oil; yield: 80%; 1H NMR: δ = 1.18 (t, J = 7.3 Hz, 3H, –CH3), 1.34 (t, J = 7.3 Hz, 3H, –CH3), 2.70 (q, J = 7.3 Hz, 2H, CH2), 3.06 (q, J = 7.3 Hz, 2H, CH2), 7.11 (s, 1H, CH), 7.20–7.90 (m, 10H, Ar–H); 13C{1H} NMR: δ = 14.2 (CH3), 14.9 (CH3), 27.2 (C, CH2), 28.1 (C, CH2), 127.5–130.5 (6Car), 133.0 (CCH), 136.0–139.2 (2Car), 139.4 (C, CH), 164.5 (C, CN); IR: 1626 (CN), 1660 (CC); C19H21NS2 (327.52): Anal. Calcd C 69.68, H 6.46, N 4.28, S 19.58; found: C 69.40, H 6.37, N 4.35, S 19.88.
4.4.5 1,1-Bis(4-chlorophenyl)-4,4-bis(methylthio)-2-azabuta-1,3-diene 5e
Yellow powder; Yield: 25%; 1H NMR: δ = 2.25 (s, 3H, SCH3), 2.51 (s, 3H, SCH3), 6.91 (s, 1H; CH), 7.13–7.63 (m, 8H; Ar–H); 13C{1H} NMR: δ = 15.9 (C, SCH3), 17.5 (C, SCH3), 128–137.5 (12Car + CCH), 139.3 (C, CH), 160.4 (C, CN); UV–vis: 261 (8100); 381 (8600); C17H15Cl2NS2 (368.34): Anal. Calcd C 55.43, H 4.10, Cl 19.25, N 3.80, S 17.41; found; C 55.58, H 3.90, Cl 19.41, N 3.71.
4.4.6 4,4-Bis(isopropylthio)-1,1-diphenyl-2-azabuta-1,3-diene 5f
Yellow powder; Yield: 55%; 1H NMR: δ = 1.17 (d, 6H, J = 6.8 Hz, CH(CH3)2), 1.32 (d, 6H, J = 6.8 Hz, CH(CH3)2), 3.25 (sept, 1H, J = 6.8 Hz, SCH), 3.83 (sept, 1H, J = 6.8 Hz, SCH), 7.10 (s, 1H, CH), 7.10-7.75 (m, 10 H, Ar–H); 13C{1H} NMR: δ = 22.6 (CH(CH3)2), 23.2 (CH(CH3)2), 37.0 (CH(CH3)2), 37.7 (CH(CH3)2), 127.5–131.0 (5Car), 132.2–139.3 (2 Car + CS2), 141.6 (CH), 165.2 (CN); IR: 1523 (CN), 1555 (CC); UV–vis: 251 (17000), 375 (10700); C21H25NS2 (355.57): Anal. Calcd C 70.94, H 7.09, N 3.94, S 18.04; found: C 70.73, H 7.12, N 4.10, S 18.00.
4.4.7 4,4-Bis(4-thiotolyl)-1,1-diphenyl-2-azabuta-1,3-diene 5g
Yellow needles (recrystallized from ethanol); yield: 75%; mp 109 °C; 1H NMR: δ = 2.30 (s, 3H, Ar–CH3), 2.33 (s, 3H, Ar–CH3), 6.99–7.02 (m, 8H, Ar–H), 7.10 (s, 1H, CH), 7.23–7.28 (m, 10H, Ar–H); 13C{1H} NMR: δ = 21.3 (2C, Ar–CH3 ), 129.3–137.4 (24Car + CCH), 140.4 (C, CH), 165.9 (C, CN); IR: 1544 (CN); UV–vis: 256 (37900), 378 (30300); C29H25NS2 (451.65): Anal. Calcd C 77.12, H 5.58, N 3.10, S 14.20; found: C 76.98, H 5.39, N 3.17, S 14.12.
4.4.8 1,1-Bis(4-chlorophenyl)-4,4-bis(4-thiotolyl)-2-azabuta-1,3-diene 5h
Yellow powder (recrystallized from ethanol); yield: 55%; mp 149 °C; 1H NMR: δ = 2.35 (s, 3H, Ar–CH3), 2.36 (s, 3H, Ar–CH3), 6.81 (s, 1H; CH), 6.99–7.64 (m, 16H; Ar–H); 13C{1H} NMR: δ = 21.3 (C, Ar–CH3), 21.4 (C, Ar–CH3), 128.6–137.5 (24Car + CCH); 138.3 (C, CH), 162.0 (C, CN); UV–vis: 251 (27700), 387 (13200); FAB+ m/z (nature of the peak, relative intensity) 520 ([M+H]+, 100); C29H23Cl2NS2 (520.54): Anal. Calcd C 66.91, H 4.45, Cl 13.62, N 2.69 S 12.32; found C 67.08, H 4.43, Cl 13.77, N 2.64.
4.4.9 4,4-Bis(4-bromobenzenethio)-1,1-diphenyl-2-azabuta-1,3-diene 5i
Yellow needles (recrystallized from ethanol); yield: 65%; mp 144 °C; 1H NMR: δ = 6.95–7.84 (m, 18H, Ar–H) ppm; IR: 1461 (CN), 1547 (CC); UV–vis: 261 (28800), 374 (19500); C27H19Br2NS2 (581.38): Anal. Calcd C 55.78, H 3.29, Br 27.49, N 2.41, S 11.03 fond C 55.94, H 3.22, Br 27.53, N 2.37 S 11.18.
4.4.10 4,4-Bis(isopropylthio)-1,1-bis(4-isopropylthiophenyl)-2-azabuta-1,3-diene 6
Yellow needles (recrystallized from ethanol); yield: 53%; mp 57 °C; 1H NMR: δ = 1.21–1.22 (d, 6H, CH(CH3)2), 1.26–1.28 (d, 6H, CH(CH3)2), 1.31–1.39 (m, 12H, 2Ar–S–CH(CH3)2), 3.11–3.20 (m, 1H, Ar–S–CH(CH3)2), 3.22–3.34 (m, 1H, Ar–S–CH(CH3)2), 3.43–3.61 (m, 1H, Ar–S–CH(CH3)2), 3.74–3.87 (m, 1H, Ar–S–CH(CH3)2), 6.93 (1H, CH), 7.11–7.72 (m, 8H, Ar–H); 13C{1H} NMR: δ = 22.6–23.5 (8C, CH(CH3)2); 36.6–38.0 (4C, CH(CH3)2); 126.4–132.6 (12C, Car), 162.3 (C, CN); UV–vis: 256 (37900), 378 (30300); C27H37NS4 (503.85): Anal. Calcd C 64.36, H 7.04, N 2.78, S 25.46, found: C 64.48, H 6.89, N 2.87, S 25.28.
4.4.11 1,1-Diphenyl-3,4-bis(tertbutylthio)-2-azabuta-1,3-diene 7a
Orange crystals (recrystallized from hexane); yield: 60%; mp 114 °C; 1H NMR: δ = 1.39 (s, 9H, –C(CH3)3), 1.40 (s, 9H, –C(CH3)3), 5.97 (s, 1H, CH), 7.28–7.82 (m, 10H, Ar–H); 13C{1H} NMR: δ = 31.1–31.7 (6C, C(CH3)3), 44.2–46.9 (2C, C(CH3)3), 127–130 (m, 13C, Car + CH), 135.8 (C, N–CC), 137.6 (C, CN); IR: 1576 (CN), 1611 (CC); UV–vis: 256 (37900), 378 (30300); C23H29NS2 (383.61): Anal. Calcd C 72.01, H 7.62, N 3.65, S 16.72; found: C 71.83, H 7.69, N 3.60, S 16.81.
4.4.12 1,1-Bis(4-chlorophenyl)-3,4-bis(tertbutylthio)-2-azabuta-1,3-diene 7b
Orange crystals (recrystallized from hexane); yield: 54%; mp 137 °C; 1H NMR: δ = 1.35 (s, 9H, –C(CH3)3), 1.43 (s, 9H, –C(CH3)3), 5.98 (s, 1H; CH), 7.19–7.63 (m, 8H; Ar–H); UV–vis: 261 (30000), 403 (1600); C23H27Cl2NS2 (452.50): Anal. Calcd C 61.05, H 6.01, Cl 15.67, N 3.10, S 14.17; found: C 60.86, H 5.97, Cl 15.74, S 14.04.
4.4.13 4,4-Dichloro-1,1-diphenyl-3-ethylthio-2-azabut-1-ene 8
A mixture of 1.1 mmol of 4a and an excess of sodium ethylthiolate (6 mmol) in 3 mL of ethanethiol was refluxed for 4 days, then poured into water (150 mL) and extracted with diethyl ether (150 mL). The organic solution was washed two times with 50 mL of water, dried over anhydrous sodium sulfate and evaporated. The crude residue was recrystallized from ethanol. Yellow powder; yield: 64%; mp = 60 °C; 1H NMR: δ = 1.45 (t, 3H, J = 7.5 Hz, CH3), 2.67 (2qd, 2H, J = 4.0 Hz, J = 7.5 Hz, CH2), 4.74 (d, 1H, J = 6.70 Hz, CH-S) ; 5.98 (d, 1H, J = 6.70 Hz, CHCl2), 7.30–7.70 (m, 10H, Ar–H); 13C NMR: δ = 14.8 (q, J = 138.0 Hz, CH3), 23.8 (t, J = 141.0 Hz, CH2), 72.2 (d, J = 156.0 Hz, CH-S), 73.8 (d, J = 181.0 Hz, CHCl2), 127.0–138.8 (8 s, 8 Car), 170.6 (s, CN); IR: 1662 (CN); FAB+ m/z (nature of the peak, relative intensity) 338 ([M++H], 15), 340 ([M++2+H], 11), 302 ([M+–Cl], 83), 304 ([M++2-Cl], 35), 276 ([M+-HSEt], 100), 278 ([M++2-HSEt], 69); C17H17Cl2NS (338.31): Anal. Calcd C 60.36, H 5.06, N 4.14, S 9.48; found: C 60.23, H 5.00, N 4.19, S 9.61.
4.4.14 Z,E-4-Chloro-1,1-diphenyl-3-ethylthio-2-azabuta-1,3-diene 9
A solution of 1.2 mmol of 4a and 4 mL of DBU in 8 mL of toluene was stirred 17 h at room temperature, then poured into 150 mL of a 0.5 M HCl solution. The organic solution was washed two times with 50 mL of water, dried over anhydrous sodium sulfate and evaporated. Yellow oil; IR: 1664 (CC); 1626 (CN).
Major isomer: 1H NMR: δ = 1.27 (t, 3H, J = 7.30 Hz, CH3); 2.72 (q, 2H, J = 7.30 Hz, CH2); 5.52 (s, 1H, CH); 7.10–7.80 (m, 10H, Ar–H). 13C NMR: δ = 14.8 (q, J = 127.6 Hz, CH3); 27.1 (t, J = 140.5 Hz, CH2); 101.1 (d, J = 20.3 Hz, CH); 125.0–132.3 (8 s, 8 Car); 139.0 (s, CS); 170.6 (s, C=N).
Minor isomer: 1H NMR: δ = 1.35 (t, 3H, J = 7.40 Hz, CH3); 2.85 (q, 2H, J = 7.40 Hz, CH2); 5.22 (s, 1H, CH); 7.10–7.80 (m, 10H, Ar–H). FAB+ m/z (nature of the peak, relative intensity): 301 ([M+.], 37.0); 303 ([M++2], 16.4).
4.4.15 Z,E-1,1-Diphenyl-3,4-bis(ethylthio)-2-azabuta-1,3-diene 7c
A mixture of 1.2 mmol of 8b and an excess of sodium ethylthiolate (10 mmol) in 6.5 mL of ethanol was heated at 65 °C for 24 h, then poured into water (100 mL) and extracted with diethyl ether (150 mL). The organic solution was washed two times with 50 mL of water, dried over anhydrous sodium sulfate and evaporated. The oily residue was chromatographed on alumina, using a CH2Cl2-petroleum ether (50:50) mixture as the eluant. Orange oil; yield: 41%; IR: 1660 (CC); 1620 (CN).
Major isomer: 1H NMR: δ = 1.00 (t, 3H, J = 7.30 Hz, CH3); 1.20 (t, 3H, J = 7.35 Hz, CH3); 2.58 (q, 2H, J = 7.30 Hz, CH2); 2.60 (q, 2H, J = 7.30 Hz, CH2); 5.47 (s, 1H, CH); 7.20–7.80 (m, 10H, Ar–H); 13C RMN: δ = 15.9 (q, J = 126.1 Hz, CH3) ; 16.7 (q, J = 127.7 Hz, CH3); 28.3 (t, J = 140.1 Hz, CH2); 29.1 (t, J = 139.5 Hz, CH2); 112.0 (d, J = 177.5 Hz, CH); 128.0–134.0 (4 s, 4 Car); 158.9 (s, C=N).
Minor isomer: 1H NMR: δ = 1.17 (t, 3H, J = 7.35 Hz, CH3); 1.25 (t, 3H, J = 7.35 Hz, CH3); 2.40 (q, 2H, J = 7.35 Hz, CH2); 2.76 (q, 2H, J = 7.35 Hz, CH2); 5.21 (s, 1H, CH); 7.20–7.80 (m, 10H, Ar–H). FAB+ m/z (nature of the peak, relative intensity): 327 ([M+.], 73.1); 298 ([M+–Et], 79.5); 266 ([M+–SEt], 9.4).
4.4.16 1,1-Diphenyl-4,4-bis(1-piperidyl)-2-azabuta-1,3-diene 10
A mixture of 6.0 mmol of NaH, 8 mL of piperidine was stirred for 15 min at 0 °C and 20 min at room temperature. Then, 1.1 mmol of 4a was added. After 36 h at 65 °C, the mixture was poured in water and extracted with diethyl ether. The organic was washed three times with water, dried over anhydrous sodium sulphate and evaporated. The crude residue was recrystallized from ethanol. Yellow powder; yield: 45%; mp = 126 °C; 1H NMR: δ = 1.45 (s br, 6H, CH2), 1.65 (s br, 6H, CH2), 2.80 (s br, 4H, N–CH2), 3.45 (s br, 4H, N–CH2), 5.40 (s, 1H, CCH), 7.10–7.70 (m, 10H, Ar–H); 13C{1H} NMR: δ = 24.8–26.76 (4C, CH2), 50.1–50.9 (2C, N–CH2), 102.4 (C, CH), 126.5–141.2 (8 Car), 148.9 (C, CCH), 157.8 (C, CN); IR: 1574 (CN); FAB+ m/z (nature of the peak, relative intensity) 373 ([M]+, 100), 374 ([M+H]+, 90), 289 ([M–NC5H10]+, 39); C25H31N3 (373.25): Anal. Calcd C 80.39, H 8.37, N 11.25; found: C 80.90, H 8.53, N 11.45.
4.4.17 [{Cu2Br2(p-iPrSC6H4)2CN–C(H)C(SiPr)2}2] 11
To a suspension of CuBr (143 mg, 1 mmol) in dichloromethane (10 mL) was added (554 mg, 1.1 mmol) of ligand 6. The mixture was refluxed for 24 h, and then the solution was allowed to reach room temperature. The filtrate was concentrated to 5 mL. After the addition of diethyl ether, the filtrate was stored at −20 °C affording red crystals. Yield: 60%; UV–vis: 257 (36900), 378 (26200); C54H74Br2Cu2N2S8 (1294.6): Anal. Calcd C 50.10, H 5.75, N 2.16, S 19.81; found: C 49.89, H 5.44, N 2.11, S 19.70.
4.5 Crystal structure determinations
Single crystals of 5i, 7a, 7b, 10 and 11 were mounted on a Nonius Kappa Apex-II CCD diffractometer equipped with a nitrogen jet stream low-temperature system (Oxford Cryosystems). The X-ray source was graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) from a sealed tube. The lattice parameters were obtained by least-squares fit to the optimized setting angles of the entire set of collected reflections. Intensity data were recorded as ϕ and ω scans with κ offsets. No significant intensity decay or temperature drift was observed during data collections. Data were reduced by using DENZO software [56]; the missing absorption corrections were partially compensated by the data scaling procedure in the data reduction. Absorption correction was further applied by using MULTISCAN [57]. The structures were solved by direct methods with SHELXS97 [58a] or SIR [58b] programs. Refinements were carried out by full-matrix least-squares on F2 using the SHELXL97 program on the complete set of reflections [58a]. All non–hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms were placed in calculated positions and included in final refinement in a riding model with the isotropic temperature parameters set to Uiso(H) = 1.2Ueq(CH), Uiso(H) = 1.3Ueq(Caromatic) and Uiso(H) = 1.5Ueq(CH3).
Crystallographic data (cif files) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers 835275 (5i), 835276 (7a), 835277 (7b), 889646 (10) and 941426 (11). Copy of these data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 IEZ, UK, fax: 144-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk.
Acknowledgement
The Ministère de la Recherche et de la Technologie, la Région de Franche-Comté and the CNRS are gratefully thanked for financial support.