

1 Introduction
Owing to the large availability of glycerol as major side-product of biodiesel industry, development of new processes to upgrade this by-product and to find new ways to valorize it is of great interest for industry [1–4]. Glycerol has been used for the synthesis of several value-added products as acrolein, dihydroxyacetone or lactic acid as few examples. One of the most widely known is the synthesis of epichlorohydrin developed by Solvay which has reached the industrial scale [5]. Glycerol carbonate as derivative of glycerol appears to be a key multifunctional chemical employed as solvent, additive, monomer, and chemical intermediate [6]. The large availability of glycerol carbonate through the multigrams scale transesterification of glycerol in the presence of dimethylcarbonate (DMC) is particularly attractive in a context of industrial applications [7,8]. In addition, owing to the bio-based origin and wide reactivity of glycerol carbonate due to the free hydroxyl or the carbonate group, this molecule is a very promising building block for organic synthesis. A large number of organic compounds have therefore been synthesized by functionalization of glycerol carbonate (GC) to produce functionalized glycerol carbonate such as esters [9–11] or ethers [12,13]. However the current synthesis of glycerol carbonate ethers essentially involves waste producing reactions. We were more particularly interested in synthesizing new carbonate glyceryl ethers according to an atom economical process which would be more compatible with the green chemistry concepts. For this purpose, we targeted to setup the metal catalyzed telomerization of 1,3-butadiene – a long-standing approach for functionalization of unsaturated substrates [14] - with glycerol carbonate in order to alkylate the remaining free hydroxyl group and introduce alkadienyl chains that can be easily hydrogenated in alkyl chains [15]. The thus synthesized carbonate glyceryl ethers with unsaturated and saturated chains are potent monomers for Ring Opening Polymerization (ROP). However, if the homopolymerization of five-membered cyclic carbonates by ROP is known to occur only at high temperatures, it is invariably accompanied with loss of CO2 [16–18]. On the other hand, it has been –scarcely– reported that copolymerization of ethylene carbonate (EC) with lactones may be an effective way to avoid decarboxylation [19], provided that it was conducted under smooth experimental conditions [20–24]. Since this approach was not studied yet with substituted five-membered cyclic carbonates as potential co-monomers, we aimed to study the ability of functionalized GC in that context, in association with ε-caprolactone [25] and lactide [26], which are the two main monomers used to prepare biodegradable polyesters by means of ROP processes. We describe herein the telomerization of 1,3-butadiene with glycerol carbonate to yield octadienyl glycerol carbonate (OGC) as well as the corresponding hydrogenated derivative, and we further present selected results that we obtained in the copolymerization of OGC with ε-caprolactone and rac-lactide.
2 Experimental
2.1 Materials and methods
All experiments were done using Schlenk techniques or a glove box (Jacomex). Pd(acac)2 and PPh3 (Strem), Al(OiPr)3 (Aldrich) and 1,3-butadiene (Linde) were used without purification. Triethylamine (Aldrich) was distilled over KOH. Toluene was purified through alumina column (Mbraun) and stored on molecular sieves in a glove box. E-caprolactone (CL) (Aldrich) was dried over calcium hydride, distilled twice, and stored over molecular sieves in a glove box. Rac-lactide (Aldrich) was purified in two steps by dissolution/evaporation from dry toluene, followed by sublimation of the monomer. Glycerol carbonate [7] and Nd(O-2,6-tert-Bu2C6H3)3 [27] were synthesized as reported. NMR (1H, 13C) spectra were recorded on a Bruker Avance 300 apparatus at room temperature in CDCl3 or DMSO-d6. The chemical shifts were calibrated using the residual resonances of the solvent. The conversion of glycerol carbonate after telomerization reaction was determined on a Shimadzu 2010 GC equipped with an Equity-5 column (25 m, i.d. = 0.32 mm). Separation of the telomers was performed on a Reveleris Flash Chromatography System on a silica column (12 g), Petroleum ether/EtOAc: from 95/5 to 80/20, flow 32 mL/min, detection 254 nm. SEC analyses were performed in THF as an eluent at 40 °C (1 mL min−1) with a Waters SIS HPLC pump, a Waters 410 refractometer, and Waters Styragel columns (HR2, HR3, HR4, and HR5E), calibrated with polystyrene standards. DSC measurements were conducted under nitrogen on a Setaram 141 system at a heating rate of 10 °C min−1 from −100 °C to +100 °C using around 10 mg samples in aluminum crucibles.
2.2 Typical procedure for telomerization of 1,3-butadiene
Pd(acac)2 (11.5 mg, 0.038 mmol), PPh3 (49.2 mg, 0.178 mmol), glycerol carbonate (7.4 g, 0.0627 mol) and triethylamine (0.84 mL, 0.006 mol) were introduced in an 80 mL stainless steel autoclave under nitrogen. Liquid 1,3-butadiene (18.75 mL, 11.25 g, 0.208 mol) was transferred at −15 °C into the autoclave. Then the autoclave was heated to 80 °C and magnetically stirred for a night. At the end of the reaction, the autoclave was cooled to ambient temperature and 1,3-butadiene was degassed before opening. The reaction mixture was analyzed by gas chromatography [Conversion = 100%, Selectivity: 5% (branched), 95% (linear, OGC)]. The linear product of telomerization (octadienyl glyceryl carbonate OGC) was isolated by flash chromatography (85% yield). NMR spectral data: 1H (DMSO, 300 MHz): δ (ppm) = 1.43 (qt, CH2); 2.02 (q, 2CH2); 3.57 (qd, CH2); 3.95 (d, CH2); 4.25 (dd, OCH); 4.52(t, OCH); 4.85–5.05 (m, = CH2 + CH); 5.40–5.90 (m, olefinic signals). 13C {1H} (APT sequence, DMSO, 300 MHz): δ (ppm) = 154.99 (CO); 138.49 (CH, CHCH2); 133.80 (CH, CHCH); 126.32 (CH, CHCH); 114.93 (CH2, CHCH2); 75.52 (CH cyclic); 71.19 (CH2,CH–CH2–O); 68.72 (CH2, O–CH2–CH); 66.12 (CH2 cyclic); 32.68, 31.11, 27.78 (3CH2). HR-MS (ESI) [M+H+] calculated for C12H19O4: 227.1278; found: 227.1271.
2.3 Hydrogenation reaction of the linear monotelomer OGC
Pd/C (40 mg), the linear monotelomer OGC (2.0 g) and dry ethanol (10 mL) were introduced in a stainless steel autoclave. The reactor was filled with 30 bar of hydrogen then magnetically stirred at room temperature for 3 h. After degassing of hydrogen, the solution was filtered and evaporated to give a clear liquid (yield: 90%) which analyses were in accordance with the molecular structure of hydrogenated OGC (H-OGC). 1H NMR (CDCl3, 300 MHz): δ (ppm) = 0.84 (t, CH3); 1.23, 1.51 (s, m, 6CH2); 3.46 (td, CH2); 3.60 (qd, CH2); δ = 4.35, 4.46 (dd, t, CH2); δ = 4.78 (m, CH). 13C {1H} (APT sequence, CDCl3, 300 MHz): δ (ppm) = 155.12 (CO); 155.12 (CH cyclic); 72.18 (CH2, CH–CH2–O); 69.63 (CH2, O–CH2–CH2); 66.33 (CH2 cyclic); 31.81, 29.47, 29.37, 29.23, 25.96, 22.65 (6 CH2); 14.10 (CH3). HR-MS (ESI) [M+Na+] calculated for C12H22O4Na: 253.1403; found: 253.1403.
2.4 Polymerization procedures
In a typical polymerization, a flask was charged in a glove box with the Nd catalyst (15 mg, 20 μmol). The solvent (toluene, 4 mL) and the monomers (500 equiv. vs. initiator) were added in this order. After a given time and temperature under stirring, the viscous mixture was treated with methanol containing 2,6-di-tert-butyl-4-methylphenol (1.0 wt %) as a stabilizer. The resulting polymeric material was dried under vacuum at room temperature to a constant weight. The yield was determined by gravimetry. The amount of OGC inserted was determined by 1H NMR from the relative integration of the signal at δ (ppm) = 2.02 belonging to the opened OGC with respect to the signal at δ (ppm) = 2.30 of the polycaprolactone backbone.
3 Results and discussion
3.1 Telomerization of 1,3-butadiene with glycerol carbonate
Glycerol carbonate (GC) was prepared by transesterification of glycerol by dimethylcarbonate (DMC) according to a procedure described in the literature [7]. We aimed to use it as a nucleophile in the catalytic telomerization reaction with 1,3-butadiene. Among the known active metals to promote this reaction, palladium proved a large superiority in terms of catalytic activity and selectivity [28–30]. We found that this reaction proceeds, with 100% atom efficiency from the reactants, through the linear dimerization of two 1,3-butadiene units with simultaneous addition of glycerol carbonate as nucleophile, in the presence of a standard catalytic system based on palladium bis(acetylacetonate) and triphenylphosphine, under smooth experimental conditions [31]. The temperature was lower than 100 °C and the reaction was performed in the presence of a weak base, Et3N. These conditions were found suitable to synthesize glycerol carbonate ethers without any degradation of the carbonate group. After 17 h of reaction at 80 °C in the presence of 0.06 mol % of palladium, a complete conversion of glycerol carbonate was achieved. The low catalyst loading and availability of the starting materials make the reaction compatible with large quantities. The reaction has been for example performed at the laboratory scale with up to 50 g of starting material. The products of telomerization were obtained with 95% selectivity into the linear octadienyl ether OGC (Scheme 1, top). The mixture of branched and linear isomers has been separated by flash chromatography thus affording the pure linear isomer with 85% yield. 1H and 13C NMR spectra of OGC with full assignation are represented in Fig. 1.
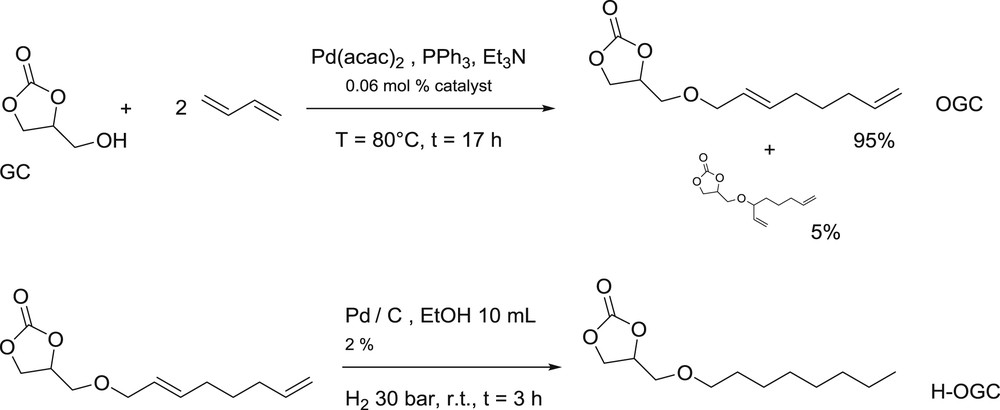
Synthesis of octadienylglycerylcarbonate (OGC) by telomerization of glycerol carbonate in presence of 1,3-butadiene (top); hydrogenation of OGC leading to H-OGC.
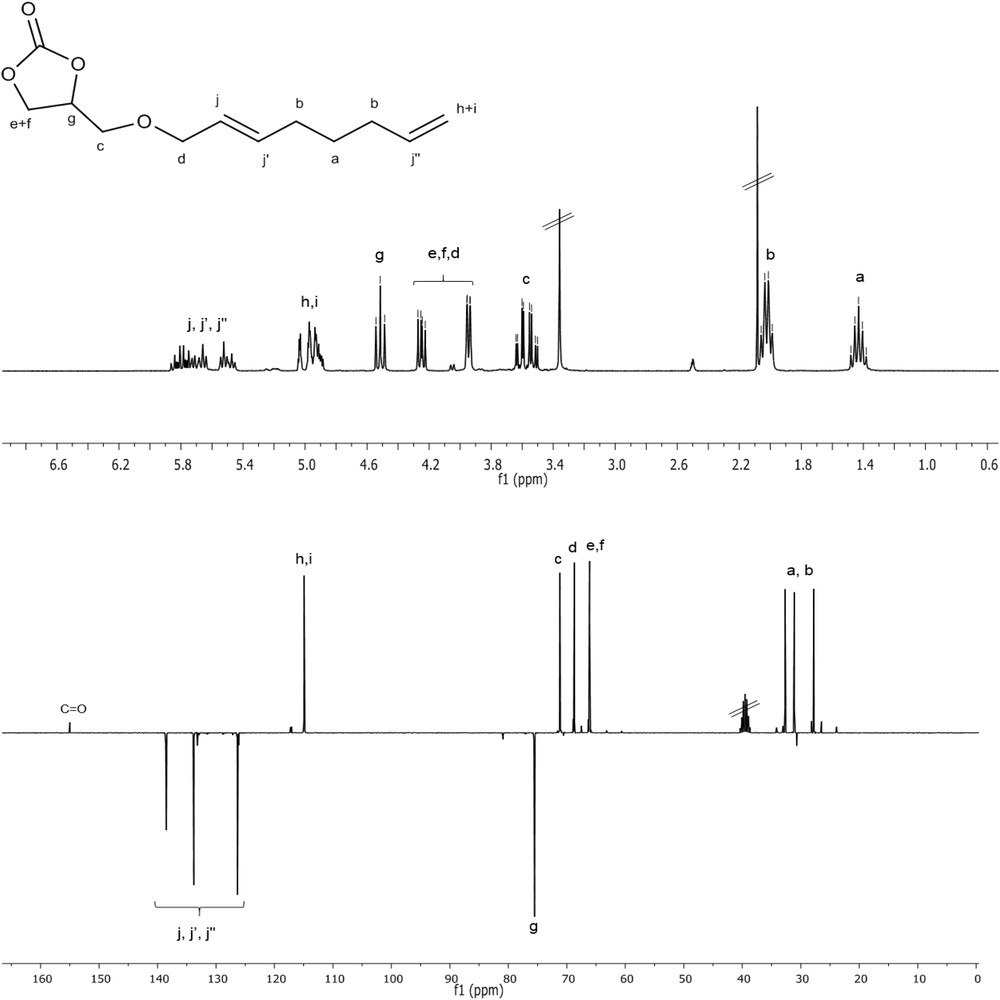
1H (top) and 13C (bottom) NMR spectra of octadienylglycerylcarbonate (OGC) with assignation (DMSO-d6).
In order to obtain more stable glyceryl carbonate ether versus oxidation reaction, the double bonds of OGC have been reduced by means of a hydrogenation reaction with Pd/C catalyst leading to hydrogenated product H-OGC in 90% yield (Scheme 1, bottom). 1H and 13C NMR spectra of H-OGC with full assignation are represented in Figure A1 (SI Section).
3.2 (Co-)polymerization experiments
Experiments were conducted with the rare earth catalyst Nd(2,6-tertBu2OC6H3)3 (here abbreviated Nd(OAr*)3), which was recently employed to insert a five -membered carbonate as co-monomer during the Ring Opening Polymerization of ε-caprolactone (CL) through the well-known coordination-insertion process [32], under smooth experimental conditions [20,23]. Representative results of copolymerization of OGC with lactones are gathered in Table 1. In a preliminary test, we checked that Nd(OAr*)3 easily homo-polymerizes ε-caprolactone (CL) under our experimental conditions (82% polymer yield, Run 1), in accordance with previous results reported [33]. By contrast, no activity was found towards OGC ring opening homopolymerization, even after prolonged time at 70 °C (Run 2). When mixtures of CL/OGC are used, the polymerization reaction is dramatically slowed down by comparison with CL alone, but it does take place at certain co-monomer ratios (Scheme 2). For an initial 70/30 (CL/OGC) monomer ratio, a polymer yield of 43% is received in 48 h at 25 °C (Run 3). As established by 1H NMR − by the shift of the c,d,e,f,g signals after opening of the ring, whereas the other resonances of the telomer moiety remained unchanged (see Fig. 2 for the numbering) − the resulting polymer material contained a little amount of OGC included (3.5%). With the same co-monomers feed but at 50 °C, the activity is better (42% in 17 h), with 3.8% molar OGC included (Run 4, Fig. 2). Increasing more the temperature did not improve this result (Run 5). At 50 °C, with higher initial CL ratio of 80 and 90 mol%, higher polymer yields were obtained (64 and 74%, Runs 6 and 7, respectively), but the amount of OGC inserted was limited to 1.5%. For 50 mol% of each co-monomer in the feed, it was necessary to carry out the reaction at prolonged time at 50 °C to recover a 15% polymer yield (Run 7). We found in that case 2.7 mol% of OGC inserted. When the OGC quantity was increased to 70 mol % in the feed vs. CL, we failed to isolate any polymeric material, despite the observation of a turbid medium at the quench with methanol after the reaction time. It is noteworthy that these low amounts on inserted five-membered cyclic carbonate co-monomer are in line with recent results, where 3–5% ethylene carbonate only were inserted in polylactide with Sn and Zn catalysts, under harsher conditions than ours [24], which confirms the difficulty of this task. The authors proposed that in the initial reaction stage lactide homopolymerization proceeds nearly exclusively, and EC incorporates to the chain as a result of recurring transesterification processes in a second stage, when most lactide has been consumed. Such mechanism can be proposed in our case, considering moreover the steric hindrance of the octadienyloxy group. We excluded to use the less sterically crowded GC instead of OGC as monomer in our tests because of the residual alcohol function, which would react with the catalyst rather than opening of the cycle. The Mn (number average molecular weight) values measured are nearly in the range expected regarding the polymer yield and the [monomer]0/[initiator] ratio of 500 used (for example ca 8000 g/mol for run 4, assuming three polymer chains per Nd metal). SEC curves often display some irregularities or shoulders, as an indication that OGC co-monomer plays a rather disturbing role during the polymerization process, but polydispersion indices Ð (defined as Ð = Mw/Mn, Mw being the weight average molecular weight) remain quite narrow (1.3–2.0 range). In comparison, with Al(OiPr)3 as initiator, it was necessary to operate at 120 °C to get significant yield, but the polymer contained 1.2 mol % of OGC incorporated (run 9).
(Co-)Polymerization of OGC with Nd(OAr*)3 as catalyst.
| Runa | [OGC]/[M] | T (°C) | t (h) | Polymer yield (%) | Mn (exp)b (g/mol) | Ðb | OGC in the polymer (%) | Tg/Tm (°C) |
| 1c | 0/100 | 25 | 0.25 | 82 | 155,000 | 1.97 | – | −61.5/58.5 |
| 2d | 100/0 | 70 | 17 | 0 | – | – | – | – |
| 3c | 30/70 | 25 | 48 | 43 | 13,000 (m) | 2.00 | 3.5 | −36.6/50.9 |
| 4c | 30/70 | 50 | 17 | 42 | 10,000 (s) | 1.66 | 3.8 | −51.8/50.9 |
| 5c | 30/70 | 80 | 17 | 44 | 17,100 (s) | 1.65 | 2.2 | −61.1/55.3 |
| 6c | 20/80 | 50 | 17 | 64 | 14,800 (m) | 1.76 | 1.5 | −36.4/57.6 |
| 7c | 10/90 | 50 | 17 | 74 | 49,100 (m) | 1.79 | 1.5 | −48.3/59.0 |
| 8c | 50/50 | 50 | 48 | 15 | 12,900 (s) | 1.33 | 2.7 | −60.7/55.2 |
| 9c,g | 30/70 | 120 | 48 | 54 | 10,200 | 1.39 | 1.2 | −60.9/58.1 |
| 10e | 30/70 | 25 | 17 | 22 | 16,600 | 1.29 | 0.8 | ndh |
| 11e | 30/70 | 50 | 17 | 30 | 16,200 | 1.29 | 1.0 | ndh |
| 12f | 10/90 | 50 | 0.5 | 74 | 61,600 | 3.15 | 3.0 | −35.9/56.3 |
| 13f | 10/90 | 80 | 0.5 | 45 | 72,700 | 1.76 | 2.0 | −61.0/59.3 |
a Nd = 0.02 mmol, [total monomer]/[Nd] = 500, V(toluene) = 4 mL; OGC: octadienylglyceryl carbonate, CL: ε-caprolactone; LA = rac-lactide; H-OGC = hydrogenated functionalized glycerol carbonate.
b Determined by SEC, calibrated with polystyrene standards, s = shoulder, m = multimodal; Ð = Mw/Mn.
c M = CL.
d No reaction for a 70/30 ratio at 50 °C.
e M = LA.
f H-OGC/CL.
g Catalyst Al(OiPr)3.
h Not determined.
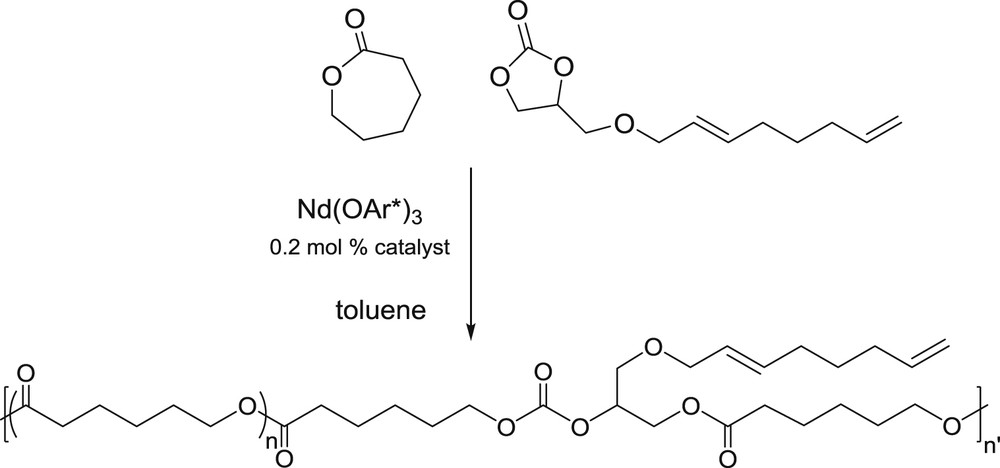
Copolymerization of OGC with ε-caprolactone.
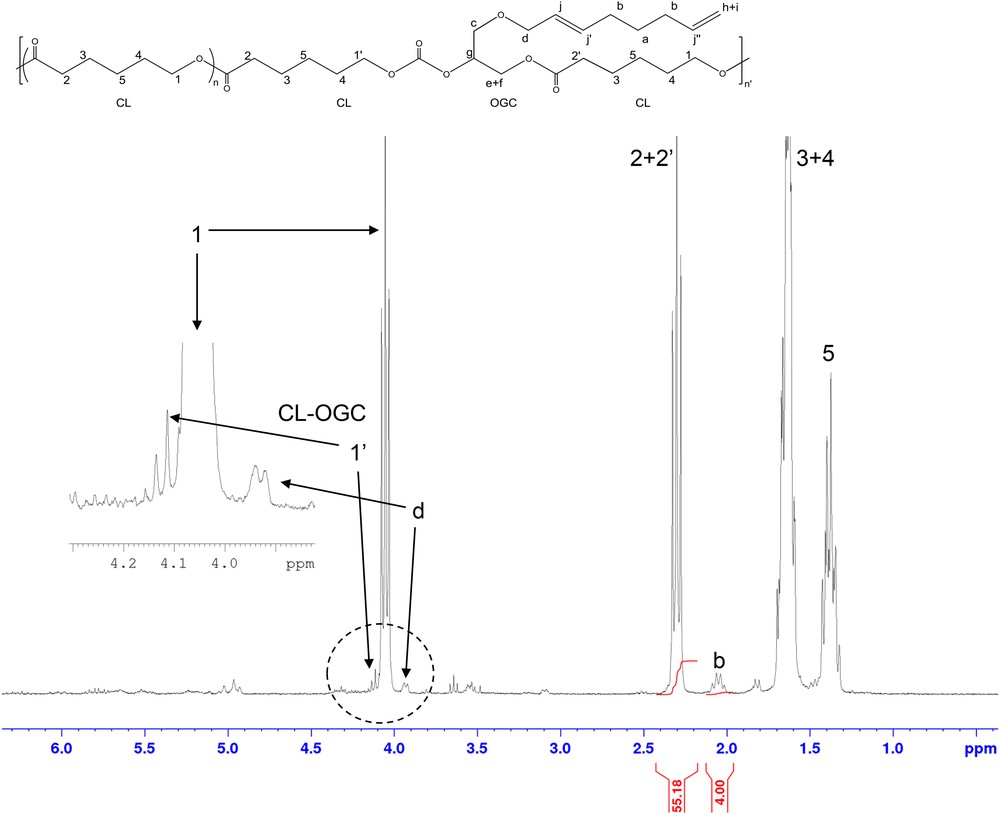
1H NMR spectrum of copolymer received from run 4 (CDCl3).
We then carried out copolymerization experiments of OGC with rac-lactide similarly as conducted with CL. After work-up, some polymeric material was isolated in all cases but very little OGC percentage (maximum 1 mol %, Runs 10, 11) was inserted in the recovered polymer, whatever the experimental conditions used. SEC curves were this time monomodal, with narrow polydispersion indices. A lower perturbing effect of the OGC co-monomer towards rac-LA than towards CL polymerization could be thus advanced. Finally, we noted that it was possible to copolymerize CL with the hydrogenated telomer H-OGC: with 10 mol % of the latter in the feed, polycaprolactone containing up to 3% co-monomer was obtained (Runs 12, 13). DSC analyses were carried out on several representative samples of copolymers isolated. We noted significant differences with the homopolymer for samples having more than 1.5% OGC co-monomer inserted (Table 1), which can be related to the amount and also the distribution of carbonate co-monomer in the polylactone backbone, as already noticed [34]. The more noticeable effects are observed for copolymers obtained from runs 3 and 4, which are the highest in OGC: the Tm is shifted of ca. 8 °C (50.9 vs 58.5 °C), and the Tg increases substantially as well, in both cases, which is typical of internal plasticization. Representative thermograms can be seen in Figure A2 (SI Section). One can therefore anticipate some modulation of the thermo-mechanical properties of the polymer by including OGC co-monomer as same as observed when α-olefins are inserted, with content of that range, into trans-polyisoprene chains [35]. Moreover, since OGC displays an unsaturated extremity, this offers further possibilities of easy and controlled chemical post-functionalization, including cross-linking, which can be of interest for specific biomedicine applications with lactone-based materials [36,37]. Further experiments are in progress to improve the insertion rate of bio-sourced OGC and OGC-H in polylactones and related biodegradable polymers and study the resulting materials in terms of performance and characterization, along with polymerization mechanisms.
4 Conclusion
The telomerization reaction with 1,3-butadiene has been used to synthesize new unsaturated and saturated glyceryl carbonate ethers from bio-sourced glycerol carbonate. The reaction is atom economical and proved to be very efficient and straightforward with low palladium loadings (0.06%) thus opening the possibility of synthesizing the monomer at higher scales. The thus synthesized linear glycerol carbonate ethers were co-polymerized with ε-caprolactone and rac-lactide by means of a neodymium initiator. Up to ca. 4 mol % of inserted carbonate monomer were obtained in the case of the copolymerization with ε-caprolactone. Such amount was enough to subtly modify the thermal behavior of polycaprolactone. Further experiments aiming at exploring the potential of the functionalized glycerol carbonate for organic and material applications are in progress.
Acknowledgments
The Region Nord-Pas-de-Calais and the University of Lille 1, Science & Technology are gratefully acknowledged for the funding of the work (MS). We also thank Aurélie Malfait for SEC measurements and Dr S. Georges for his help in DSC measurements.