

1 Introduction
Among the alternatives for energy production, solar conversion presents itself as the most promising in terms of sustainability and durability. There are two main strategies to use solar power as a source of energy: (1) converting light energy directly to electricity through the use of photovoltaics [1,2] or, (2) the more promising scenario, storing light energy in the form of chemical bonds [3–11]. In Photosystem II, nature uses sunlight to strip electrons and protons from water in order to do chemical synthesis [12–17]. This process has inspired the field of artificial photosynthesis, where the challenge is to duplicate the principles of light capture, generation of charge-separated states, charge accumulation, and catalytic formation of high energy content products using simpler molecular devices. Different approaches can be conceived in order to use light to drive the oxidation of water. One way is to couple photovoltaic devices with molecular catalysts capable of oxidizing water to O2 [10,18,19], while another is to design a molecular photocatalyst containing both a photoactive and a catalytic module to achieve direct photoelectrochemical water oxidation [19–21]. The goal in the latter strategy is to accumulate sufficient oxidizing equivalents at a catalytic center through intramolecular light-driven electron transfers to drive the oxidation of water. Styring and co-workers [22,23] were the first to propose such a model based on ruthenium trisbipyridine covalently bound to a manganese complex. Wieghardt and co-workers [1,24] have also studied light induced electron transfer in a RuII–MnIV complex resulting in the formation of a MnIV–phenoxyl radical state. The inherent advantage of such structural motifs in terms of circumventing diffusion-limited chemistry has led to the elucidation of details such as the Marcus inverted region [3,5,7,9], and the role of energetics in studies of long range electron and energy transfer [12,14,25]. However, a challenging task within these supramolecular assemblies is to underpin the mechanism of the excited state reactivity. The operating mechanistic pathways often depend on a variety of factors including the electronic structures of the constitutive modules and the degree and nature of the electronic communication between them. Recently [18,19], we have developed an extended, rigid molecular spacer designed to hold both the photoactive unit and a putative catalytic core. We have explicitly shown that our molecular spacer that contains an imidazole motif behaves as a redox active module [19,26]. In the present study, our goal was to expand this chromophore-relay complex in order to create a chromophore-relay-catalyst complex. Terpyridine ligands were used as organic scaffolds to host the metal ion for the catalytic unit. In this study, we report on the corresponding MnII derivative. Reasons behind this choice reside in the fact that dinuclear manganese complexes from this family of ligands have been reported to perform catalytic reactions (Brudvig's catalyst [22,25] for both the oxidation of water and organic substrates [24]). It is to be noted that Najafpour and coll. have clearly evidenced the formation of manganese oxide nanoparticles that act as the real catalyst stemming from this di-μ-oxo dinuclear manganese compound for the water oxidation reaction [27].
Herein, we report the synthesis and characterization of a series of ruthenium polypyridine-based complexes covalently bound to a terpyridine ligand, which is capable of coordinating a MnII ion (Scheme 1). We have studied the effect of varying the redox properties of the photoactive module by including ester groups on the bipyridines. Intramolecular electron transfer between the photogenerated RuIII and the MnII metal ions was analysed on the basis of DFT calculations. For simplicity we abbreviated the mononuclear ruthenium complexes as 1a and 2a while their corresponding RuII–MnII derivatives are 1b and 2b, respectively.

Synthesis of mononuclear (1a, 2a) and binuclear (1b, 2b) complexes.
2 Results and discussion
The organic framework designed to hold both metal centers presents two metal binding sites, 1,10-phenanthroline to bind the ruthenium ion and a 2,2′:6′,2″-terpyridine intended to lodge the manganese ion (see Scheme 1). Given the presence of two metal coordination sites in our target ligand, the synthetic pathway whereby the ligand is prepared prior to metal insertion was discarded in order to avoid indiscriminate metallation processes. Instead, we have used a synthetic tactic where a modified Ru-polypyridine complex serves as a molecular platform to initiate the construction of the target molecules (see Scheme 1). This synthetic approach relies on the inert nature of the low spin RuII-polypyridine complex, which prevents demetallation or ligand exchange under the reaction conditions. RuII heteroleptic complexes containing either bipyridines or ethyl ester-modified bipyridines together with a coordinated phendione ([(bpy)2Ru(phendione)]2+ and [(bpy(CO2Et)2)2Ru(phendione)]2+) were prepared. These complexes were then reacted via a Steck and Day reaction with ether, 4′-formyl terpyridine in refluxing acetic acid, and in the presence of an excess of ammonium acetate [28–30]. After purification, the target mononuclear RuII salts were isolated in good yields (>70%). Insertion of the MnII ion into the terpyridine cavity was performed in a mixed solvent solution following modified literature procedures [25].
2.1 UV-visible spectra
The ground state absorption spectra for the series of complexes, compounds 1a (Ru–IT), 2a (estRu–IT), 1b (Ru–IT–Mn) and 2b (estRu–IT–Mn) in acetonitrile, are shown in Fig. 1. Compound 1a shows spectral features comparable to those of the parent [Ru(bpy)3]2+ compound, with the π–π* transitions at around 280 nm and the double-hump metal to ligand charge-transfer (MLCT) transitions peaking at 450 nm. An additional band at 320 nm is observed, which was previously assigned [19,31] to an intra-ligand charge transfer (ILCT) transition for the imidazole containing ligand. The ester derivatives 2a and 2b display a 25-nm red shift of the MLCT band (peaking at 475 nm) and a redshift by about the same amount of the π–π* transition (Fig. 1). The presence of the MnII ion in the terpyridine cavity does not influence the intensity of the MLCT band of the RuII core, which tails up to 600 nm. However, the presence of Mn appears to induce a redshift of the 320–nm absorption band to 345–355 nm, suggesting a complex origin of this absorption feature. Of note is that the model tpy–MnII (IT–Mn) or the non-metalated ligand shows no electronic features in the MLCT region.

UV-visible absorption spectra in acetonitrile for 1a, 1b, 2a, and 2b. Due to poor solubility no extinction coefficient was determined for 2b. The spectrum of the non-metalated ligand phenImtpy (in DMSO) is shown for comparison.
2.2 Electrochemistry
The redox potentials for all the compounds studied were obtained in CH3CN and 0.1 M TBAPF6 and are given in Table 1. The cyclic voltammogram of the mononuclear complex 1a shows the three consecutive one-electron reduction waves ranging from −1.41 to −1.96 V, which are typical features of RuII polypyridine-type complexes. On the anodic side of the CV, 1a presents a first irreversible wave prior to the quasi-reversible metal-centered RuIII/II couple, which is attributed to the oxidation of the imidazole ring. The irreversible nature of this wave (see Fig. S1 in the Supporting information) is probably linked to a proton coupled electron transfer process. The redox potential for the RuIII/II couple (1.27 vs SCE for 1a) falls within the range observed for polypyridine RuII complexes [19,26]. The shift of the cathodic waves by about +250 mV for both compounds 2 is evidence of the effect of the electron-withdrawing ester groups on the periphery of the bipyridine skeleton. This effect is also perceptible on the oxidation potential of the RuII centre which was shifted to more positive potential by ∼250 mV. The oxidation potential of the imidazole unit is also modified in a similar fashion. The electrochemical behaviour of the bimetallic Ru–Mn complexes 1b and 2b is characterized by a quasi-reversible wave in the range of 1.00–1.11 V versus SCE. This wave is assigned to the one electron oxidation of the MnII ion, by analogy to the oxidation of the mononuclear tpyMnCl2 (IT-Mn) complex run under the same conditions [19,25]. In fact, no noticeable shift in the redox potentials of the manganese metal center is observed in the dinuclear complexes. If we consider that the driving force (ΔG°) for electron transfer from the Mn metal to the photo-oxidized chromophore is given by the difference between the half-wave potentials of the two metal centers [E1/2(MnIII/II) – E1/2(RuIII/II)] then the introduction of the ester groups leads to a ∼ (220–280) mV increase in driving force for the photo-oxidation reaction (Table 1).
Electrochemical data for complexes (1 mM) in acetonitrile.
| Compound | E1/2a RuIII/II | E1/2a Im+/Im | E1/2a MnIII/II | E1/2a bpy-type ligands | ΔG°b [eV] |
| (1a) Ru–IT | 1.27 | 1.22 | −1.41, −1.61, −1.96 | −0.05 | |
| (1b) Ru–IT–Mn | 1.30 | 1.09 | −1.41, −1.61, −1.96 | −0.21 | |
| (2a) estRu–IT | 1.51 | –c | −1.01, −1.24, −1.73 | ||
| (2b) estRu–IT–Mn | 1.54 | 1.11 | −1.01, −1.24, −1.73 | −0.43 |
a Determined by CV at 100 mV s−1; E1/2 = 1/2 (Epa + Epc) in V versus SCE in the presence of tetra-n-butylammonium perchlorate.
b ΔG° = −FeΔE1/2, and ΔE1/2 calculated as the difference between E1/2(RuIII/II) and the lowest oxidation potential [E1/2(Im+/Im) or E1/2(MnIII/II)].
c Imidazole oxidation not observed in the presence of ester groups.
2.3 Photochemistry
2.3.1 Emission
Data on the luminescence emission are collected in Table 2. The emission maximum for the complex 1a occurs at around 610–620 nm, similar to that of the parent [Ru(bpy)3]2+ complex, while the emission maximum for the ester derivatives 2 is observed around 640–650 nm. The presence of the MnII ion in the terpyridine cavity does not affect the wavelength of the emission maximum indicating that the energy level of the emissive 3MLCT state of the chromophore is not modified. Luminescence quantum yields and lifetimes for the mononuclear ruthenium complexes 1a and 2a are reported in Table 2. The emission kinetics are characterized by a monophasic decay with ca. 1.6 μs lifetime. The presence of MnII in the terpyridine cavity leads to a quenching of the emission (Table 2). We observed that in acetonitrile the emission decay of the dinuclear RuII-MnII complexes shows a biphasic behavior, characterized by a short-lived component (50 ns) and a long-lived one (1.0 μs) (See Fig. S2 in the Supporting information). The relative amplitude of the two phases varies between the two dinuclear complexes such that the amplitude of the short-lived component decreases for the ester derivative 2b compared to 1b. Both kinetic phases show the same emission spectrum (see Fig. S3 in the SI for data for complex 1b) and are similarly quenched upon addition of an external electron acceptor (vide infra). This excludes the possibility of the fast phase being an artifact. Furthermore, it is worth noting that the lifetime of the long-lived component for the dinuclear RuII–MnII complexes does not correspond to the lifetime of the analogous mononuclear RuII complexes, thus ruling out that this component might stem from a possible de-coordination of the MnII ion from the terpyridine cavity. A possible explanation for the origin of the biexponential decay could be the presence of two different quasi-degenerate excited states, spectrally indistinguishable within the given experimental precision. This interpretation will be further discussed below.
Luminescence data in acetonitrile for the mononuclear ruthenium complexes and the dinuclear ruthenium-manganese complexes.
| Compound | λmax [nm] | ΦL | A1 | τ1 [ns] | A2 | τ2 [ns] |
| [Ru(bpy)3]2+ | 610 | 0.059a | 1 | 860 | – | – |
| 1a Ru–IT | 618 | 0.073 | 1 | 1650 | – | – |
| 2a estRu–IT | 640 | 0.051 | 1 | 1600 | – | – |
| 1b Ru–IT–Mn | 617 | 0.003 | 0.15 | 1000 | 0.85 | 50 |
| 2b estRu–IT–Mn | 640 | 0.015 | 0.85 | 690 | 0.15 | 120 |
a ΦL [Ru(bpy)3]2+ [26].
2.3.2 Transient absorption
The transient absorption spectrum obtained upon excitation at 460 nm for the complex 1a is similar to that of [Ru(bpy)3]2+ and is characterized by the depletion of the MLCT ground state absorption around 460 nm accompanied by the appearance of a weak absorption above 500 nm, extending into the near IR region (see Fig. S4 in the Supporting information). For the ester derivative 2a, the maximum of the depletion is red-shifted, in agreement with the shift in the absorption spectrum. For the mononuclear complexes, the kinetics of absorption decay correspond to the kinetics of the emission. In the case of the dinuclear Ru–Mn complexes, the transient absorption shows a biphasic decay with lifetimes close to those found in the emission measurements.
2.3.3 Photoinduced electron transfer in the presence of an external acceptor
Light induced electron transfer experiments were performed in the presence of methyl viologen (MV2+) as a reversible electron acceptor. For the mononuclear RuII complexes (1a and 2a), the bimolecular rate constants for the interaction between the complexes and the electron acceptor (MV2+) were determined from studies of the emission lifetime as a function of the concentration of MV2+. The analysis of the corresponding Stern-Volmer plots yields a bimolecular rate constant of about 2 × 109 M−1 s−1, a value which is in agreement with data for related systems. Concerning the dinuclear RuII–MnII complexes, both phases of the emission kinetics were similarly quenched by MV2+.
In Fig. 2, kinetic traces of absorption changes at 470 and 605 nm in acetonitrile are displayed for the complexes studied. These traces are used as spectroscopic probes for the recovery of the RuII from oxidized RuIII and for the presence of the reduced state of the electron acceptor, MV+, respectively. For all complexes, the depletion signal at 470 nm decays faster than the absorption signal at 605 nm indicating that intramolecular electron transfer occurs. For the mononuclear complexes 1a and 2a the difference is small, indicating a slow rate constant kIET for an internal electron transfer of 4.5 and 6.2 × 103 s−1 for 1a and 2a, respectively. In these cases, the imidazole ring is the only redox center that may account for the recovery of the RuII state.
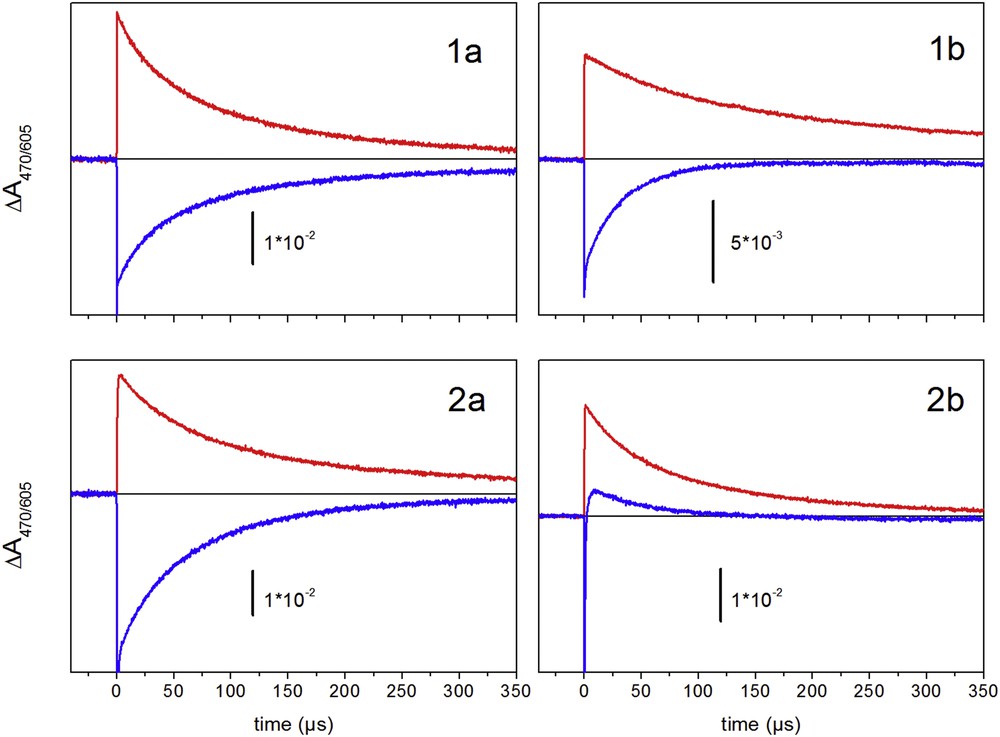
Absorption kinetics at 605 nm (positive) and at 470 nm (negative) in the presence of MV2+(PF6-)2 upon excitation at 450 nm in acetonitrile. Solutions were purged with argon for 15 min before measurements.
For the bimetallic complexes, the kinetic analysis of the absorbance transients at 470 and 605 nm (Fig. 2, right), yielding rates of 7.0 × 103 (1b) and 1 × 106 s−1 (2b) for the recovery of RuII by internal electron transfer. In these complexes the electron donor to RuIII could be imidazole or the manganese ion. The absence of distinct absorption features in the UV/vis related to MnIII/II transition does not allow us to detect Mn oxidation by absorption change data.
To verify Mn oxidation, EPR measurements were performed on degassed solutions of the dinuclear RuII–MnII complexes, subjected to continuous light excitation (>450 nm, xenon lamp) in the presence of an irreversible electron acceptor and then quenched in liquid nitrogen. Before this treatment, all the dinuclear RuII–MnII complexes are characterized by the typical six-line spectrum of high spin MnII centers at g ∼2.00. Fig. 3 shows the EPR spectra before and after irradiation for 1b and 2b. In the case of 1b, after irradiation at λ longer than 435 nm for 2 min in the presence of a diazonium salt as a sacrificial electron acceptor, the MnII signal completely disappeared. Upon longer illumination times, no trace of a different multi-line signal arising from the formation of higher oxidation states of MnIII–MnIV μ-oxo dimers was detected. Under our experimental conditions, we could not observe the signal for a MnIII complex in the EPR parallel mode probably due to the weak concentration of the photogenerated MnIII species and the less sensitive parallel mode EPR spectroscopy. Similar EPR experiments were performed on the bimetallic complex incorporating ester groups (2b) in the presence of [CoIII(NH3)5Cl]2+ as an electron acceptor (Fig. 3, right). The six-line signal observed in the dark disappears under illumination with the concomitant formation of the signal of CoII at g = 4.44, leading to the same evidence for photo-oxidation of MnII.
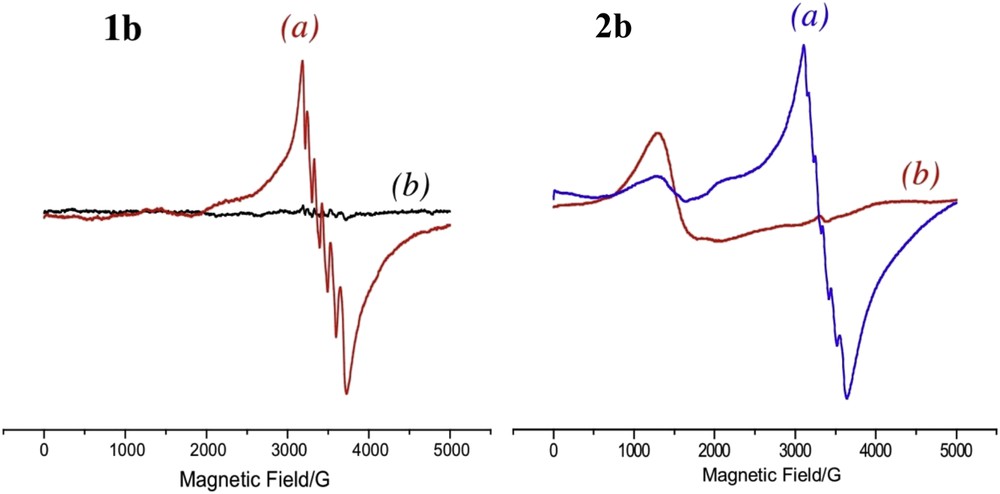
Left: X band EPR spectra of a mixture of 1b with diazonium salt before illumination (a), and after illumination (b). Right: X band EPR spectra of a mixture of 2b and cobalt pentamine before illumination (a), and after illumination (b). Experimental conditions: microwave frequency 9.415 GHz; microwave power 3.196 mW; modulation amplitude 25.251 G, temperature 5K, time constant 81.92 ms; scan width 5000 G; modulation frequency 100 kHz.
3 Theoretical results
DFT calculations were performed on selected compounds (1a, 1b and 2a) in order to rationalize their photophysical behavior. These DFT analyses are also based on theoretical results gained from the study of a previously reported RuII complex fused with a rigid phenanthroline and an imidazole ligand [(bpy)2RuphenImPh]2+ [19,26]. In this report, we have gathered in the Supplementary information all the studies on the mononuclear complexes 1a and 2a, in their ground state S0, the lowest triplet state T1, and the lowest D0 doublet state of the oxidized form. We have also performed DFT calculations on a model complex with MnII/III in the terpyridine cavity, termed IT–Mn, for the subsequent interpretation of the electron transfer processes in the bimetallic complex (1b). The optimised geometries of the manganese cavity appropriate for MnII and MnIII are abbreviated as [A] and [B] respectively in the text. We also indicate the stepwise approach to model these large molecular systems.
Recent theoretical studies on ruthenium-polypyridine complexes have pointed out the importance of taking into account solvent effects in order to improve the description of their electronic [28–30,32], and photophysical properties [31]. We have analyzed all our chemical systems in solution in the framework of the polarizable continuum model (PCM) [33]. Following our previous studies, we have chosen water as the solvent medium.
3.1 Mononuclear ruthenium complexes
The presence of ester groups in the complex 2a has striking effects: the HOMO-LUMO gap δ is smaller and is responsible for the red shift observed for the MLCT absorption band (Fig. 1). Moreover, stabilization of the d orbitals is consistent with the increased RuIII/II oxidation potential in this complex.
The excited triplet energy for complexes 1a and 2a lies, according to DFT calculations, at 2.32 and 2.01 eV above the ground state respectively, in agreement with the trend observed for the emission maxima occurring at 618 and 640 nm. The corresponding luminescence lifetimes are longer than that of the parent [Ru(bpy)3]2+ complex but comparable to those of the previously reported analogous family with a fused phenanthroline and imidazole rigid ligand [19]. The inherent reason for the increased lifetime is not known; however, according to DFT calculations, the intervention of excited states from the conjugated rigid ligand could be the origin for such behavior.
Photoinduced electron transfer experiments in the presence of MV2+ show similar decay kinetics at 470 nm and 605 nm for complex 1a, suggesting slow intramolecular electron transfer (Fig. 2). These results are in agreement with DFT calculations, which indicate that oxidation takes place on the ruthenium. On the other hand, the faster recovery at 470 nm indicates the occurrence of an intramolecular electron transfer in complex 2a. Based on the electrochemical data obtained for compounds 1a and 2a, the available driving force to promote the oxidation of the imidazole containing ligand by the photogenerated RuIII is almost identical for both mononuclear compounds. As we reported previously, intramolecular electron transfer between an imidazole motif and photogenerated RuIII is a function of the protonic state of the imidazole fragment [19]. We rationalize the difference in the electron transfer processes between 1a and 2a as originating from a less basic imidazole ring in the case of 2a as compared to 1a. We argue that in the excited states of compound 2a, the deprotonation of the imidazole ring is more facile, hence leading to faster oxidation of the imidazole ring by RuIII.
3.2 Mono-oxidized dinuclear Ru–IT–Mn complex
In Fig. 4, we gather the main outcomes of our computed data. All calculated states of 1box are drawn to scale. Schematic Marcus-type parabolas have been added to connect the same state in the two geometries. The energy difference of 0.26 eV between the bottom of the two wells represents the parameter ΔG° from the Marcus equation. This value compares rather well with the energy difference 0.21 eV calculated from the RuIII/II and MnIII/II oxidation potentials obtained by electrochemistry (see Table 1). The results gained from calculations on 1box allow us to conclude that light absorption by 1b in the presence of an electron acceptor is likely to generate a RuII–MnIII state after geometry relaxation.
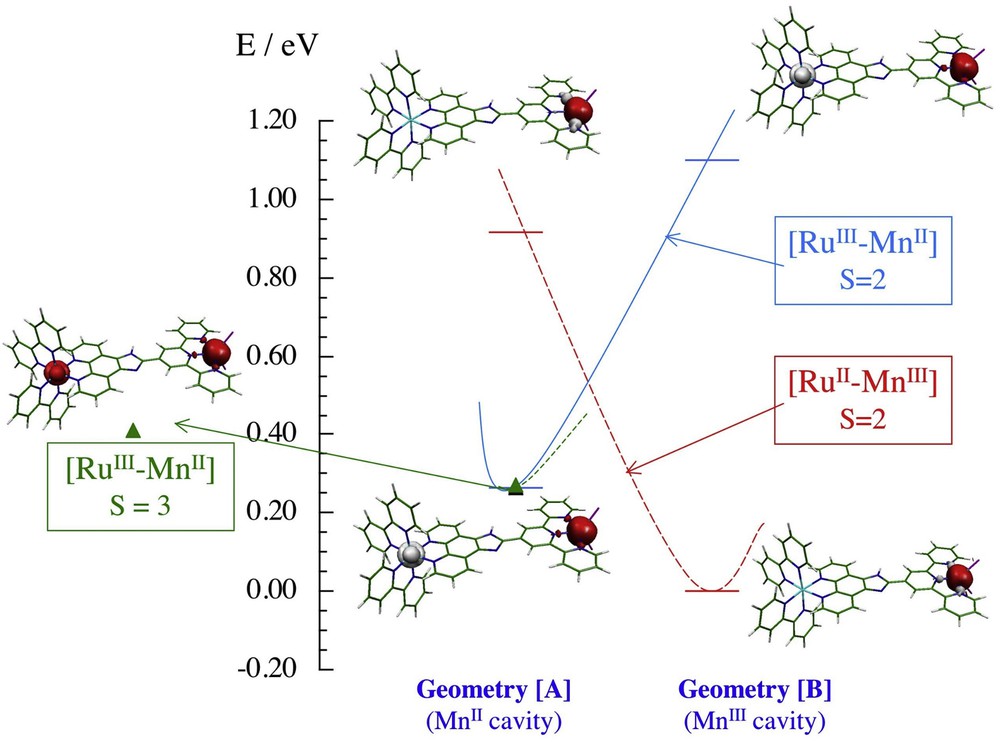
Schematic diagram of the S = 3 and S = 2 states of 1box calculated in the local geometry of the MnII ion (geometry [A]) and of the MnIII ion (geometry [B]). The bars represent computed energies and the corresponding spin density distributions are presented. Marcus-type parabola correlates each state in the two geometries.
3.3 Photoinduced electron transfer
In this section, we analyse the physical and functional properties of the investigated complexes in the light of the theoretical results obtained for compounds 1a, 2a and 1b. In the presence of the reversible electron acceptor MV2+, experimental results obtained for all the bimetallic complexes indicate that the photochemically produced RuIII is reduced to the original RuII oxidation state upon oxidation of the coordinated Mn(II) ion in the terpyridine ligand. EPR studies in the presence of an irreversible electron acceptor ([CoIII(NH3)5Cl]2+ or diazonium) support the change in the oxidation state of the manganese ion. An important aspect in this research is to understand how the intramolecular electron transfer progresses.
The recovery of the RuII from the photo-oxidized RuIII in the ester derivative, 2b, is much faster than that of the other complexes (Fig. 2). In addition, a transient positive absorption appears in the 470 nm traces. We interpret this result as being due to the presence of a transient reaction product formed by an internal electron transfer to RuIII from a redox couple different from MnIII/II, because the MnIII shows no absorption in this spectral region. This oxidized state then decays on a slower timescale by recovering an electron from MnII. The obvious candidate for the intermediate electron donor is the imidazole group. This interpretation is supported by the observation of an intramolecular electron transfer between the photogenerated RuIII and the imidazole in the presence of an electron acceptor for compound 2a. Thus, it is very likely that this electron transfer pathway is also operating for the dinuclear complex 2b although no distinct electrochemical process could be distinguished on the cyclic voltammograms. The proposed route for the electron transfer processes for compound 2b is depicted in Eq. 1.
| (1) |
Of note, for compound 1b, only a monophasic decay is detected, presumably because the slow oxidation of the imidazole group does not allow for build-up of detectable amounts of the radical intermediate.
4 Conclusions
We have reported here the synthesis of a novel family of heteroditopic ligands capable of binding to a photoactive chromophore (RuII polypyridine) and also serving as a molecular platform that can host a MnII ion. The synthetic route we have established provides an easy way to modulate the intermetallic distance and also the redox properties of the photoactive unit. The laser flash photolysis experiments were performed in the presence of external electron scavengers (MV2+ or [CoIII(NH3)5Cl]2+). For the dinuclear RuII–MnII complexes (1b and 2b) we observed the formation of the strongly oxidizing [RuIII(bpy)3]3+ moiety, which is then reduced by an intramolecular electron transfer process. For compound 1b, a monophasic decay describing an electron transfer to the RuIII from the MnII takes place and is described by a rate constant of k ≈ 104 s−1, leading to the formation of MnIII. The value of this rate constant falls in the range of electron transfer rates for previously reported RuII–MnII systems. The slow electron transfer rate found in these synthetic models is mainly attributed to the large reorganization energy around the manganese center. Interestingly, we found that for the ester-bearing derivative (2b), the recovery of the RuII from the photogenerated RuIII is much faster (k ≈ 106 s−1). We visualize that the electron transfer process from the MnII to RuIII in this case proceeds through a rapid electron transfer from the imidazole moiety to the RuIII followed by a slower positive charge shift to the MnII ion. This is an important result in the construction and design of photocatalysts where the imidazole moiety can act as an electron relay between the photoactive unit and the putative catalyst. Such an integrative unit in a supramolecular sensitiser–catalyst assembly can help to synchronize fast photoinduced electron transfers with slow redox chemical reactions.
5 Experimental section
5.1 Materials and methods
Acetonitrile and methanol (Aldrich) were of HPLC grade. [Ru(bpy)3]Cl2 (Aldrich), methyl viologen MV(PF6)2 (Aldrich), and [CoIII(NH3)5Cl]Cl2 (Aldrich) were used as received. Tetrabutyl ammonium perchlorate (Aldrich) was used as the supporting electrolyte in electron transfer experiments as well as for electrochemical measurements.
5.1.1 Electrochemical measurements
Cyclic voltammetry was performed with an EGG PAR (model 273 A) electrochemical workstation. Acetonitrile was distilled prior to each experiment and the solution (1 mM for the complexes, 0.1 M of tetrabutyl ammonium perchlorate) was introduced into an argon-purged electrochemical cell. The experiments were performed at room temperature using a glassy carbon electrode as the working electrode (3 mm2 surface area), a platinum wire as the counter electrode, and a Ag/AgClO4 (0.01 M) electrode in acetonitrile as the reference electrode (+0.292 V vs SCE).
5.1.2 Mass spectrometry
Mass spectra were recorded on a Finnigan MAT95S by direct injection of a mixture of CH2Cl2/MeOH/H2O (1/1/1). Nuclear magnetic resonance experiments were performed on a Bruker Avance 400 spectrometer (400.13 MHz).
5.1.3 EPR measurements
X-band electron paramagnetic resonance spectra were recorded on a Bruker 300 spectrometer at 5 K with the following parameters: microwave 3 mW, modulation amplitude 25 G, time constant 80 ms, and modulation frequency 100 kHz. Photodriven oxidation was performed by mixing the required Ruthenium compounds (CH3CN, 0.5 mL, 1 mM) with [Co(NH3)5Cl]Cl2 (H2O, 2.5 mL, 20 mM) resulting in solutions 0.17 mM in Ruthenium complex and 17 mM in electron acceptor. The samples were degassed with argon and illuminated with an 800 W tungsten lamp for 10 min with both infrared and UV (400 nm) cutoff filters. After this time, 100 μL aliquots were transferred into a quartz tube, which was immediately immersed into liquid nitrogen for EPR analyses.
5.1.4 UV-visible-NIR
Spectra were recorded on a Specord 210 (Analytic Jena) spectrophotometer. Values for the molar extinction coefficients (ε, M−1 cm−1) were obtained from Beer–Lambert plots by varying the sample concentration between 10 and 250 μM. Linear dependences were obtained for all samples in this concentration range.
5.1.5 Photochemistry
Luminescence spectra were recorded on a Varian Cary Eclipse spectrofluorimeter. Luminescence quantum yields (FL) were obtained for optically matched, diluted (A(λexc) < 0.1) solutions upon excitation at 460 (MLCT maximum) and 532 nm (laser excitation wavelengths) by comparing the area under the spectrum, using [Ru(bpy)3]2+ as the reference [26]. Emission lifetimes were measured using a frequency-doubled nanosecond Nd/YAG laser (532 nm, duration 7 ns, 2 mJ cm−2, Quantel). The detection wavelengths were selected with interference filters (610 or 650 nm, ∼10 nm width) and signals were detected with a micro-channel plate photomultiplier tube (R2566U, Hamamatsu) and recorded using a digital oscilloscope (TDS 744 A, Tektronix). Laser flash-absorption transients were recoded with an Edinburgh Instruments flash photolysis system (LP920) consisting of a 450 W pulsed xenon lamp for measuring light, a monochromator (Czerny–Turner) and a silicon photodiode as the detector. Signals were amplified with a wideband preamplifier (model 5185, EG&G) before recording with a digital oscilloscope (TDS 3034B, Tektronix). The sample was excited at 90° to the measuring beam by a flash from a frequency-tripled Nd/YAG laser (Surelite, Continuum) equipped with an OPO (SLOPO, Continuum). Excitation was set at 450 nm (duration 5 ns, 2 mJ cm−2). In the electron transfer experiments, MV2+ was used as a reversible electron acceptor. For all optical measurements 10 × 10 mm sealed cuvettes were used and samples were purged with argon for 10 min prior to measurements.
5.1.6 Computational methods
Density Functional Theory calculations were carried out using Becke's three-parameter hybrid functional B3LYP [34,35] along with the valence double-z basis set LanL2DZ [36,37] including the Los Alamos effective core potential for heavy atoms. For the nonclosed-shell state, calculations were of unrestricted type. All the calculations were performed using the Gaussian 03 [38] software package. The solvent medium was taken into account in the framework of the polarized dielectric model (PCM) [33,39] using the integral equation formalism (IEF) [40,41]. The selected solvent was water and all parameters were kept to their implemented Gaussian 03 values.
Geometry optimizations were performed for the closed shell (phenImtpy) and (phenImPhtpy) ligands as well as for the manganese complex [phenImtpyMnCl2] both in its neutral (IT–Mn) and mono-oxidized (IT–Mnox) forms. For the ruthenium complex 1a, the interpretation of the photophysical experiments, in the absence as well as in the presence of an electron acceptor, may involve the lowest triplet state T1 and the ground doublet state D0 of the oxidized complex. However, as these complexes are very large ones, no geometry optimizations were attempted and the molecules in the ground state S0 were constructed with the aid of previous results. This holds a fortiori for the bimetallic complex [(bpy)2RuphenImtpyMnCl2]2+ (1b) comprising 96 atoms. Details are reported in the supporting information section case by case.
5.2 Synthesis and product characterization data
The bipyridine ligands as well as the [Ru(bpy)2(Cl)2] were synthesized following modified literature procedures.
5.2.1 [(bpy)2Ru(phendione)](PF6)2 (3)
484 mg of Ru-bipyridine dichloride (1 mmol, 1 equiv) were reacted with 338 mg of silver nitrate (2 mmol, 2 equiv) in methanol for 2 h. The solution was filtered in order to remove the silver salt, and the filtrate was evaporated under reduced pressure. The solid was re-dissolved in ethanol and one equivalent of phendione (1) (210 mg, 1 mmol) was added. The solution was refluxed in the dark for three hours. At this time the solvent was evaporated, the remaining solid re-dissolved in a minimum amount of methanol, and the desired compound was precipitated by drop wise addition of a saturated aqueous solution of sodium hexafluorophosphate and chromatographed in alumina using acetonitrile as the eluent. (275 mg, 44%). 1H-NMR. (400 MHz, CD3CN): 8.56–8.49 (m, 6H), 8.10 (t, 3J = 5.6 Hz, 4 H), 7.98 (dd, 3J = 5.6 Hz, 4J = 1.4 Hz, 2H), 7.86 (d, 3J = 5.2 Hz, 2H), 7.75 (d, 3J = 5.6 Hz, 2H), 7.63–7.59 (m, 2H), 7.43 (t, 3J = 5.6 Hz, 4H) ppm. MS (ESI) = 312.1 [M]2+, 624.1 [M]+, 769.1 [M+PF6]+.
5.2.2 [(bpy(CO2Et)2)2Ru(Phendione)](PF6)2 (4)
Synthesis of this compound was done following the above procedure using 4, 4′-diethyl ester bipyridine. (65% yield). 1H-NMR. (400 MHz, CD3CN): δ 9.08 (dd, 3J = 5.3 Hz, 4J = 1.7 Hz, 4H), 8.55 (d, 3J = 5.9 Hz, 2H), 8.08 (d, 3J = 5.8 Hz, 2H), 7.96–7.91 (m, 3H), 7.89 (dd, 3J = 5.9 Hz, 4J = 1.7 Hz, 2H), 7.84 (dd, 3J = 5.3 Hz, 4J = 1.7 Hz, 2H), 7.69–7.53 (m, 2H), 4.46 (q, 3J = 7.8 Hz, 8H), 1.41 (t, 3J = 8.0 Hz, 12H) ppm. MS (ESI) = 456.1 [M]2+, 1057.1 [M+PF6]+.
5.2.3 [(bpy)2RuphenImtpy](PF6)2 (1a)
83.78 mg of terpyridine-4′ carbaldehyde 5 (0.321 mmol, 1 equiv) were reacted with 200 mg [(bpy)2Ru(phendione)]2+ (2) (321 mmol, 1 equiv) in 10 mL acetic acid. The mixture was heated to 80 °C for 30 minutes. At this time 495 mg of dry ammonium acetate (6.42 mmol, 20 equiv) were added and the reaction mixture was heated in the dark at 100 °C for 12 h. The solvent was evaporated under reduced pressure and redissolved in a minimum amount of MeOH. An aqueous solution of NaPF6 was added drop wise and the resulting precipitate was filtered, washed with cold MeOH and dried under vacuum. Purification by column chromatography (neutral alumina, 95:5 Acetonitrile: Methanol) afforded 224 mg of product, 81% yield.
1H-NMR. (400 MHz, (400 MHz, CD3CN): δ 9.31 (s, 2H), 9.04 (d, 3J = 8.2 Hz, 1H), 8.97 (d, 3J = 8.3 Hz, 1H), 8.79 (d, 4J = 4.6 Hz, 2H), 8.65 (d, 3J = 7.9 Hz, 2H), 8.55 (dd, 3J = 14.8, 4J 8.2 Hz, 4H), 8.21–8.08 (m, 4H), 8.03 (t, 3J = 7.8 Hz, 2H), 7.95 (td, 3J = 7.7, 4J = 1.8 Hz, 2H), 7.87 (d, 3J = 5.5 Hz, 2H), 7.82 (dd, 3J = 8.3, 4J = 5.3 Hz, 2H), 7.64 (t, 3J = 6.7 Hz, 2H), 7.49–7.45 (m, 4H), 7.34–7.18 (m, 2H) ppm. MS (ESI) = 1010.1 m/z, [M+PF6]+, 432.7 m/z, [M]2+.
5.2.4 [(bpy(CO2Et)2)2RuphenImtpy](PF6)2 (2a)
Synthesis of this compound was done by reacting terpyridine-4′ carbaldehyde 5 with 4 and following the same procedure as that for 1a. (72% yield).
1H-NMR. (400 MHz, DMSO): δ 9.34 (m, 4H, bpy-H), 9.20 (m, 2H, tpy-H), 8.84 (d, 3J = 5.58 Hz, 2H, phen-H), 8.75 (d, 3J = 7.75 Hz, 2H, tpy-H), 8.65 (m, 4H, bpy-H), 8.26 (d, 3J = 7.72 Hz, 2H, tpy-H), 8.11 (m, 2H, tpy-H), 7.97 (m, 2H, phen-H), 7.87 (s, 4H, bpy-H), 7.73(t, 3J = 7.53 Hz, 2H, phen-H), 7.51 (m, 2H, tpy-H), 4.45 (q, 3J = 7.1 Hz, 8 H, CH2H), 1.39 (t, 3J = 7.5 Hz, 12H, CH3H) ppm. MS (ESI) = 1298.2 m/z, [M+PF6]+, 576.2 m/z, [M]2+.
5.2.5 [(bpy)2RuphenImtpyMnCl2](PF6)2 (1b)
Compound 1a (52 mg, 0.060 mmol, 1 equiv) was dissolved in a minimal amount of acetonitrile. To this solution, 75 mg of MnCl2 (0.60 mmol, 10 equiv) dissolved in a minimal amount of MeOH were added and the solution was stirred in the dark under an Ar atmosphere at room temperature. The precipitate formed was filtered and washed with MeOH and ether. The resulting red/orange powder was redissolved in MeOH and precipitated by adding some drops of a saturated aqueous NH4PF6 solution. This mixture was filtered, washed with MeOH and ether, and dried under vacuum. The reaction yielded 46 mg of product. (77% yield). MS (ESI) = 495.3 m/z, [M]2+.
5.2.6 [(bpy(CO2Et)2)2RuphenImtpyMnCl2](PF6)2 (2b)
This compound was obtained following the same procedure as that for compound (1b). (64% yield). MS (ESI) = 634.1 m/z, [M]2+.
Acknowledgements
This work was supported by ANR TechBioPhyp (ANR 2010 BLAN 0926 02), the LABEX CHARMMMAT (ANR-11-LABX-0039), and by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01. A. G. T thanks the Chateaubriand for a scholarship.