

1 Introduction
Exploration of chemosensors for bivalent cations, particularly iron and copper, is an important research subject attracting widespread attention due to the pivotal roles of such species in several biological processes [1,2]. A lack/surplus of these two trace essential elements in the human body can induce some serious diseases such as cancer and system perturbation [3,4].
Although many analytical techniques were used to detect metal cations including high performance liquid chromatography (HPLC), fluorescence, inductively coupled plasma mass spectrometry (ICP-MS) and atomic absorption spectrometry (AAS) [5], this field still suffers many performance limitations such as analysis time and high cost and needs sophisticated equipments. For all these reasons, the scientific community devoted huge interest to develop new electrochemical sensors, which are considered economical, easy to handle, and highly sensitive systems [6].
Ferrocene and rhodamine were widely used as electrochemical mediators to sense metallic cations. For instance, P. Beer et al. employed a polyaza ferrocene macrocyclic ligand to sensitively sense copper (II) ions by the formation of a stable ferrocene based electrochemical complex in polar organic solvent or even in water [7]. Moreover, R. Ziessel's group has explored complexation properties to detect various metals such as Cu (I), Cu(II), Fe(II) and Co(II) [8,9]. Furthermore, rhodamine was also frequently involved in metallic cation detection. Recently, dimethyliminocinnamyl based rhodamine has been explored by Kamal et al. as an electroactive compound to detect iron (Fe2+) [10]. More recently, rhodamine hydrazide derivatives have also been used as a highly sensitive and selective copper (Cu2+) electrochemical sensor in aqueous solution [11]. However, relatively few examples of phosphine chalcogenides have been described as complexation agents for the detection of metal cations [12–17]. Indeed, Bis-(diphenylthiophosphoryl)ferrocene (dptpf) and seleno analogue (dpspf) were considered as the ferrocenyl analogues of tetraphenylimidodiphosphinate ligands Ph2P(E)-NH-P(E)Ph2 (E = S, Se) [14] and their bonding ability towards Cu(I) was reported [15]. Furthermore, the (C5H4P(S)(NMe2)2)2Fe and (C5H4P(S)(NEt2)2)2Fe have been developed by Nifant'ev and co-workers [16] to stabilize different metallic cations [17].
In this work, we report the use of bis(tetraethylthiophosphoramidoyl)methylamine, MeN[P(S)(NEt2)2]2, as an electrochemical chemosensor to detect Ca2+, Ni2+, Cd2+, Co2+, Cu2+ and Fe2+ bivalent cations and to sense simultaneously Fe2+ and Cu2+. The electrochemical investigations reveal drastic changes in the potential of the oxidation peak of the ligand in the presence of above cited metallic cations. Furthermore, the presence of Fe2+ and Cu2+ in the same media induces a simultaneous anodic shift in the potential of the oxidation peak. The electrochemical results were confirmed by the UV–Visible technique where we noticed a redshift behavior.
2 Materials and methods
2.1 General
All reactions were carried out under a nitrogen atmosphere in solvent dried by standard techniques and stored over activated 3 Å molecular sieves. All reagents and solvents were of analytical reagent grade, purchased from Sigma-Aldrich and Acros and used without further purification. Cadmium (II) perchlorate hydrate Cd(ClO4)2·6H2O, copper (II) perchlorate hydrate Cu(ClO4)2·6H2O, cobalt(II) perchlorate hydrate Co(ClO4)2·6H2O, iron (II) perchlorate hydrate Fe(ClO4)2·6H2O, nickel(II) perchlorate hydrate Ni(ClO4)2·6H2O and calcium (II) perchlorate hydrate Ca(ClO4)2·4H2O were used as received.
The NMR spectra were recorded on a Bruker AC-300 MHz spectrometer. Chemical shifts for 1H and 13C NMR were referenced to tetramethylsilane and 31P was referenced to 85% H3PO4 in D2O. UV–Visible spectra were measured in 1 cm quartz cells using a UNICO Spectro-Quest 2800 spectrophotometer. The IR spectrum was obtained on a Perkin–Elmer alpha ATR apparatus.
2.2 Electrochemistry
The electrochemical experiments were conducted at ambient temperature in 0.1 M tetrabutylammonium perchlorate in acetonitrile solution with a three-electrode glass cell controlled by MetrohmAutolab PGSTAT101 electrochemical workstations. The cell was fitted with platinum wire as the counter electrode and a platinum electrode as the working electrode (1 mm diameter) which was polished before each experiment. The Ag/AgCl (3 M KCl) electrode was used as the reference electrode. Metal cations were added as solutions of the corresponding perchlorate salts in anhydrous acetonitrile.
2.3 Preparation of chemosensor MeN[P(S)(NEt2)2]2
MeN(PCl2)2 was obtained by the reaction between methylammonium chloride and the excess of phosphorus trichloride. MeN(PCl2)2 (11.26 g, 4.8 10−2 mol) was added dropwise to a solution of Et2NH (40 ml, 0.38 mol) in anhydrous diethyl ether (150 ml) cooled at −78 °C. The reaction mixture was stirred for 10 h at −78 °C. After filtration, the obtained oil was dissolved in toluene (250 ml) and an equivalent amount of sulfur (2.35 g, 7.210−2 mol) was added at 0 °C. The desired compound was obtained by recrystallization from light petroleum as a yellow solid. 31P{1H} NMR(CDCL3) δ/ppm: 75.18 (s). 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.8 (t, 3H, (CH3N)); 3.2 (q, 16H, CH2); 1.1 (t, 24H, CH3). 13C NMR (CDCl3, 75.1 MHz) δ/ppm: 26.4, 15.1, 40.9. IR: υ(PNP) 932 cm−1, υ(PN) 1168 cm−1 and υ(PS): 709 cm−1. GC–MS (t = 17.83 min): m/z = 443 [M]+.
3 Results and discussion
3.1 Synthesis of electroactive ligand L: MeN[P(S)(NEt2)2]2
Reported chemosensor L was prepared as presented in Scheme 1 according to a previously published procedure [18]. Furthermore, the ligand structure was confirmed by spectroscopic techniques such as 31P, 13C and 1H NMR and GC–MS analysis.
Schematic route of synthesis of ligand L.
3.2 Electrochemistry of free ligand L
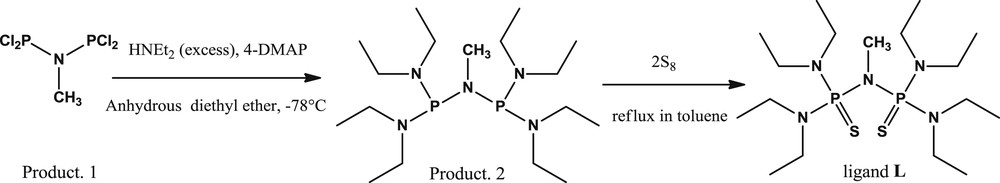
The electrochemical properties of electroactive ligand L were determined by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in anhydrous acetonitrile using 0.1 M TBAClO4 as a supporting electrolyte. In fact, the cyclic voltammogram of L exhibits a quasi-reversible electrochemical signal presented by an oxidation peak which is attributed to the oxidation of chalcogen moiety. The sweep back displays a reduction peak related to the reformation of the neutral ligand [18]. Compared to previous studies describing the electrochemical studies of different mono and bisphosphine sulfides, title ligand L displays important changes in the electrochemical characteristics, especially in terms of reversibility [19].
Furthermore, the linear dependence of the logarithm of the oxidation peak current ipa on the logarithm of the scan rate with a slope close to 0.50 indicates a diffusion-controlled electrochemical process [20] (Fig. 1A). Differential pulse voltammetry was explored due to its high sensitivity and resolution compared to other electrochemical techniques [21]. DPV of free ligand L shows the presence of an oxidation peak at 0.58 V (Fig. 1B).
(A): Cyclic voltammograms of free chemosensor L solution (1.0 mM) at different scan rates from 100 to 500 mV/s in 0.1 M TBAClO4 acetonitrile solution. Inset: Variation of the logarithm of oxidation peak current with the logarithm of scan rate. (B): Differential pulse voltammogram of free chemosensor L.
3.3 Electrochemistry of metallic complexes
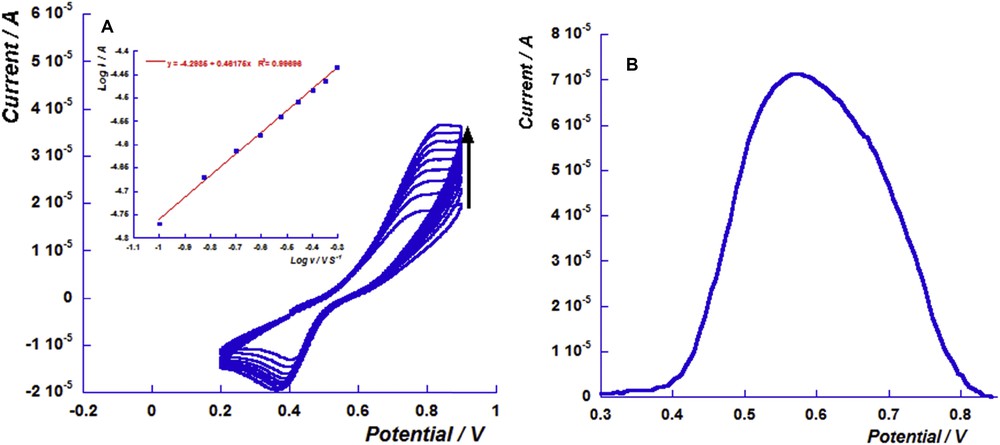
The sensing event of metallic cations (M2+: Ca2+, Cd2+, Cu2+, Ni2+, Co2+ and Fe2+) was studied and characterized by electrochemical methods such as differential pulse voltammetry, chronoamperometry and cyclic voltammetry (CV). Cyclic voltammograms presented in Fig. 2A summarize the different voltammograms of the free ligand, copper and iron complexes (For all other cations cited previously, see Fig. S.1 in ESI).
(A): Cyclic voltammograms of 1.0 mM solution of free ligand L in the presence of 0.5 equiv of iron perchlorate and copper perchlorate cations in 0.1 M TBAClO4 acetonitrile solution. Scan rate: 0.1 V s−1. (B): Curve of the ratio of the nickel perchlorate cation concentrations on the ligand concentration versus the oxidation current.
The CV results showed that the presence of metallic cations induced considerable changes in the redox properties of L. The progressive addition of each metallic cation M2+ to a solution of chemosensors led to a progressive disappearance of its oxidation peak, while a new oxidation peak was observed at a higher anodic potential attributed to the oxidation of the formed complex (Fig. 2B). In fact, the observed shift can be explained by the electronic charge delocalization from of the chalcogen moieties towards to metal centre [22,23].
As can be seen from Table 1, the anodic potential shifts were comprised between 346 and 880 mV. For instance, the potential value was at 670 mV in the presence of Ni2+ while the detection of Ca2+ induced a potential shift of 480 mV. Furthermore, the presence of iron cation Fe2+ induces the highest potential shift. To summarize, the shift value depends on the nature of bivalent cations and increases in the following order: Co2+ ∼ Cu2+ < Ca2+ < Cd2+ < Ni2+ < Fe2+.
Thermodynamic dataa of L in presence of 0.5 equiv of Ca2+, Cd2+, Cu2+, Ni2+, Co2+ and Fe2+.
| Ca2+ | Cd2+ | Cu2+ | Ni2+ | Co2+ | Fe2+ | |
| Epa(C)/V | 1.22 | 1.36 | 1.08 | 1.41 | 1.06 | 1.62 |
| ΔEpa(L)/mV | 480 | 620 | 347 | 670 | 346 | 880 |
a 0.1 M TBAClO4 in acetonitrile solution, scan rate: 0.1 V s−1; working electrode: Pt (diameter: 1 mm); Pt counter electrode and Ag/AgCl (3 M KCl) reference electrode.
It has been demonstrated that the shift value ΔE between the oxidation potential of the free ligand and its complex results from the polarizing power of the metallic cations [24,25]. The important potential shifts, observed after the chelation process, indicate a strong interaction between the S-donor binding sites and metallic centre. In fact, the charge of the two sulfur atoms confirms the effect of their involvement in the complexation process [26]. Therefore, we conclude that bis(tetraethylthiophosphoramidoyl)methylamine is a good chelating agent for various bivalent metallic cations due to the presence of a chelate ring ensuring the stability of the formed complexes [27,28].
In addition, DPV studies show that the disappearance of the oxidation peak of L was complete when 0.5 equiv of the nickel cation was added (Fig. 2B), suggesting the formation of the 1:2 metal-ligand complex [Ni(L)2]2+. The 1:2 stoichiometry of the formed complex was also observed in the case of calcium(II), cobalt(II) and cadmium(II) cations (See Figs. S.2–4). Moreover, these results were confirmed by chronoamperometry as a complementary technique which exhibited the stabilization of the current complex beyond 0.5 equiv (See Figs. S.5–8).
To further confirm the 1:2 stoichiometry of formed complexes, mass spectrometry was carried out to deduce with high precision the mass of the complexes. The recorded spectra show the presence of various peaks localized at 942, 498 and 443 in the case of the iron complex and 946, 506 and 443 in the case of the copper complex related to the mass of [ML2]+, [ML]+ and [L]+ fragments, respectively.
3.3.1 Sensing of iron bivalent cation Fe2+
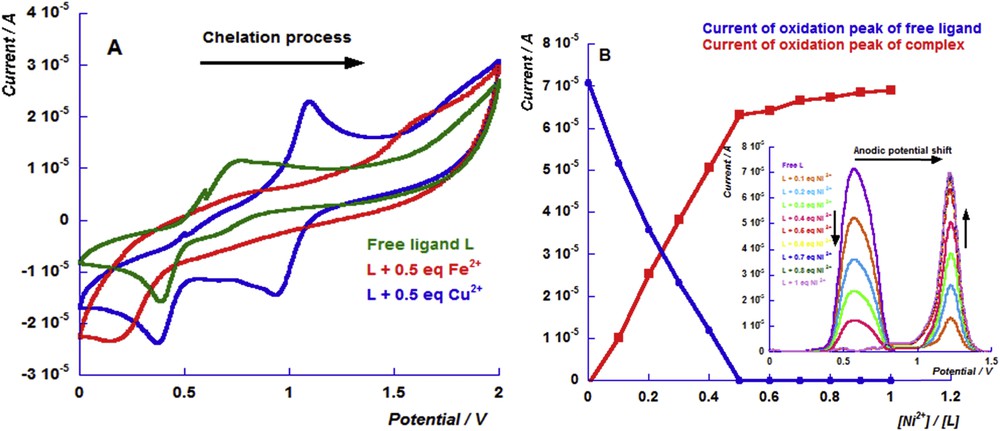
The sensing process of iron cations induced a huge change in the redox properties of free chemosensor L. In fact, the cyclic voltammogram of [Fe(L)2]2+ shows an important anodic shift reaching 880 mV, which indicates the high stability of this complex (red curve in Fig. 2A). The successive additions of different equivalents of iron cations induce a progressive increase in the oxidation peak current of the iron complex, accompanied by a considerable decrease in the oxidation peak current of free ligand until its disappearance (Fig. 3A). The conversion was complete when 0.5 equiv of iron cations was added. Furthermore, fixing the applied potential at the oxidation peak potential of the iron complex, chronoamperogram results confirm those obtained by DPV experiments. The increase in the current was found to be 32.24 μA upon addition of 0.2 equiv of Fe2+. Beyond 0.5 equiv, the variation of the current value becomes low (Fig. 3B).
(A) Differential pulse voltammograms of the successive additions of the iron perchlorate cation (from 0.1 to 1 equiv) to the free ligand L solution (1 mM) in 0.1 M TBAClO4 acetonitrile solution and (B) the chronoamperograms of the addition of the iron perchlorate cation equivalents to the L solution (1 mM) performed on the oxidation potential of the iron complex.
3.3.2 Sensing of copper bivalent cation Cu2+
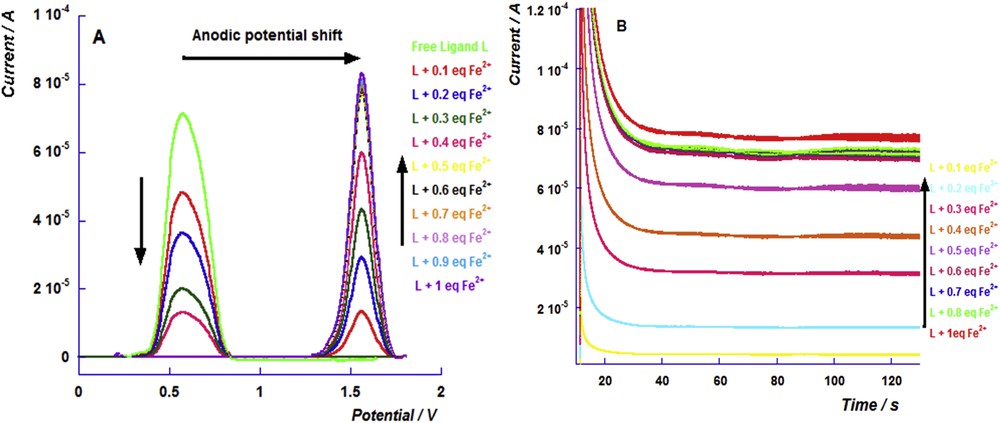
The copper sensing triggered a considerably lower anodic shift (347 mV) than that induced by the iron chelation. Indeed, the copper chelation with the thiophosphoryl ligands led to a weak lengthening of the PS bond [15]. Fig. 4A depicts the differential pulse voltammograms of the successive additions of different equivalents of Cu2+. Similar to the results shown above, we noticed an increase in the current of the oxidation peak of the copper complex and a decrease in that of the free ligand. The chronoamperograms show that the current of the oxidation peak of the copper complex increases up to the addition of 0.5 equiv (Fig. 4B).
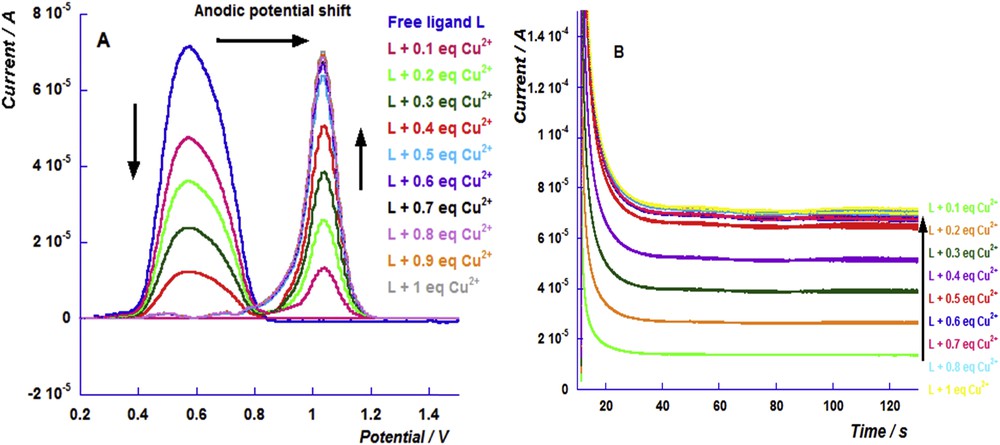
(A) Differential pulse voltammograms of the successive addition of the copper perchlorate cation to the free ligand solution and (B) the chronoamperograms of the different additions of the copper perchlorate cation (from 0.1 to 1 equiv) to the free ligand solution performed on the oxidation potential of the iron complex.
Since the oxidation peak potentials of the free ligand, iron and copper complexes are well separated, Fe2+ and Cu2+ cations were sensed simultaneously. However, the simultaneous detection of other bivalent cations was experimentally difficult due to the overlap and the approximation of potentials.
3.3.3 Simultaneous sensing of copper and iron bivalent cations
Simultaneous sensing of copper and iron cations was carried out and the results are shown in Fig. 5. The sensing of iron cation Fe2+ in the presence of 0.1 equiv of the copper cation, reaching 0.4 equiv, stabilizes the signal of iron complex oxidation (Fig. 5A). A solution of the free ligand in the presence of 0.1 equiv of iron cation Fe2+ was exposed to a series of successive additions of the copper cation. The DPV results revealed a similar behavior to that observed for a series of copper additions (Fig. 5B).
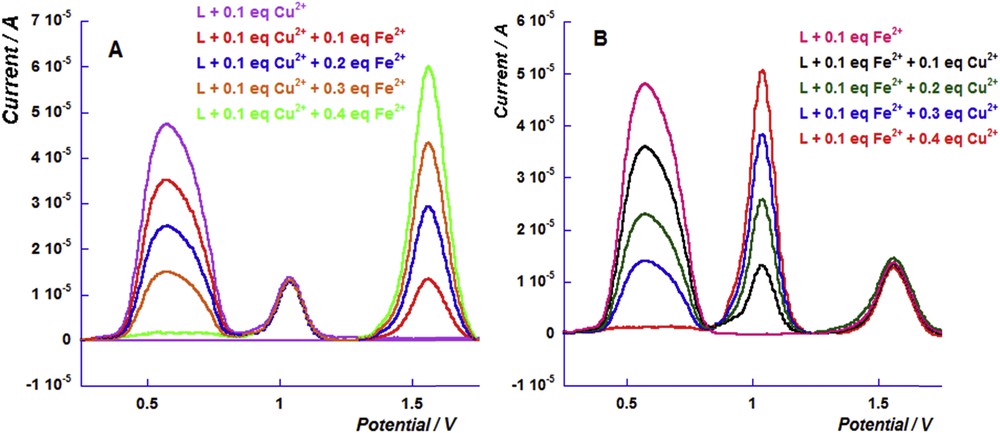
(A) Differential pulse voltammograms of the successive additions of the iron perchlorate cation to the solution of ligand L (1 mM) in the presence of 0.1 equiv of the copper perchlorate cation and (B) Differential pulse voltammograms of the successive additions of the copper perchlorate cation to the solution of ligand L in the presence of 0.1 equiv of the iron perchlorate cation.
To evaluate the performance of electroactive chemosensor (L), a comparative study was developed. Indeed, Table 2 presents various ferrocene based chemosensors, described in the literature for iron and copper cation detection [29–33]. The results of Table 2 show that ligand L presents the highest potentiometric shift values compared to those cited in the table. This is presumably due to the presence of strong S,S′ based receptor coordination sites. Furthermore, it should be noted that the title sensor does not contain or require a transducer moiety in contrast to the other chemosensors.
3.4 UV–Visible spectroscopic study
During the sensing process, a considerable change in solution color due to the formation of metallic complexes was noticed (inset Fig. 6). This color change allows the exploration of the UV–Visible spectroscopy as a control technique. The UV–Visible spectrum of the free ligand shows two absorbance bands (the major one being at 273 nm while the second one at 313 nm) which correspond to the π → π* transitions within the PS groups and the n → π* electronic transitions, respectively [34,35]. Successive additions of iron cations (Fig. 6A) and copper cations (Fig. 6B) to an acetonitrile solution of L induce a redshift and an increase in the absorption intensity of the two bands.
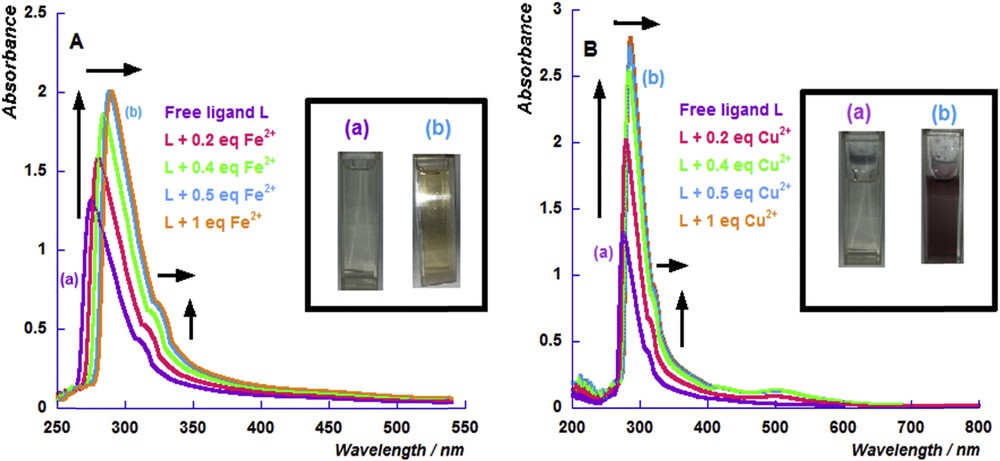
UV–Visible absorption spectra before and after the successive additions of the iron perchlorate cation (A) and copper perchlorate cation (B) to an acetonitrile solution of L.
In the case of the copper complex, the presence of a new absorption feature at 504 nm was observed, consistent with a metal-ligand charge transfer (MLCT) band of the copper (I) complex. This is due to the reducible behavior of thiophosphoryl ligands toward the Cu(II) cation. This is in fair agreement with results reported by Meek et al. who demonstrated that the copper (I) perchlorate complexes [Cu(Ligand)2]ClO4 were prepared upon reduction of copper (II) perchlorate with the diphosphine disulfide ligands (Et4P2S2 and Me4P2S2) [38]. In order to confirm this hypothesis, the UV–Visible absorbance spectrum was recorded after the addition of 0.5 equiv of Cu+ to L solution. We noticed the presence of the same characteristic bands when 0.5 equiv of Cu2+ was added to L solution (see Fig. S.9). This suggests that title ligand L is able to reduce the bivalent copper cation and forms a stable copper(I) complex, [Cu(L)2]+.
Moreover, UV–Visible absorbance spectra were recorded after the addition of 0.5 equiv of different bivalent cations cited above to L solution and showed the same behavior as described above (Fig. S.10). The observed shifts can be explained by thiophosphoryl groups to metal charge transfer, confirming the formation of desired complexes [36,37]. Up to 0.5 equiv, we noticed a stabilization of the absorption signal which clearly indicates the [M(L)2]2+ complex formation.
4 Conclusions
A new electrochemical organothiophophoryl chemosensor was explored to sense different bivalent cations such as Cu2+, Ni2+, Co2+, Ca2+, Cd2+ and Fe2+. The electrochemical results revealed remarkable anodic shifts of the oxidation peak potential of free ligand L in the presence of different cations. Extremely important oxidation potential shifts of up to 880 mV were observed upon the chelation process depending on the nature of tested cations. The 1:2 metal-ligand stoichiometry of the complexes was confirmed by electrochemical and UV–Visible techniques. The good separation and resolution of the two-oxidation peaks of iron and copper complexes were explored to simultaneously detect iron (Fe2+) and copper (Cu2+) cations.
Acknowledgements
The authors wish to thank the Tunisian Ministry of High Education and Scientific Research for the financial support (LR99ES15) of this work and Dr M.A.K. Sanhoury for technical assistance.