

1 Introduction
Direct conversion of lignocellulosic biomass (LCB) attracts increasing interests for the production of fuels and chemicals. Very recent studies have shown that liquefaction of raw LCB in usual solvents may lead to the preferential conversion of one wood component into chemicals, whereas keeping the other native wood components almost unchanged. Liu et al. [1] have described the formation of sugar alcohols in high yield from raw LCB processed in hot water in the presence of Pt/C catalyst, while the solid residue was enriched in lignin. We have also reported the possibility to obtain lactic acid directly from pinewood sawdust in hot water using ZrW catalyst, and similarly, the solid residue still contained unconverted lignin [2]. Besides, in liquid alcohols, several teams have reported the catalytic conversion of raw LCB into phenolic derivative, whereas unconverted cellulose is recovered in the solid residue [3–6]. Beyond these recent reports of selective wood fractionation and conversion in liquid media, alternative strategies using supercritical (SC) fluids have gained a growing attention because of their peculiar solvation properties and ability to weaken hydrogen bonds and, therefore, to dissolve materials that are not soluble in usual liquid solvents [7]. In this field, subcritical water has already been intensively investigated due to its efficiency to liquefy LCB [8,9]. However, the use of this environmentally friendly solvent presents certain drawbacks such as severe experimental conditions (Tc = 374 °C and Pc = 221 bar) associated with corrosion issues, which makes the process scale-up difficult. Besides subcritical and SC water, various SC organic solvents such as alcohols (i.e., methanol, ethanol, and propanol) [10–13] or acetone [14–17] have also been used to liquefy LCB. Compared to SC water, they present the advantages of milder critical conditions and better solvent ability to dissolve biomass derivatives [1,18,19]. Particularly, enhanced yields of liquid products [14,20,21] and limited solid yields were reported using SC alcohols [22,23]. Methanol and ethanol are, by far, the most studied SC alcohols applied to LCB liquefaction. In a few articles, these two light SC alcohols were compared for delignification issues [24] or for woody material liquefaction [17,19]. In general, there is an agreement on the better efficiency of ethanol in experiments performed in a batch type reactor, under isothermal conditions, which is attributed to its better solvent properties related to its lower dielectric constant (24.3 at 25 °C). Concerning the efficiency of SC ethanol treatment of an LCB for a selective liquefaction/conversion of one of its constitutive biopolymers (i.e., lignin, hemicelluloses, or cellulose), the published works are less clear. Although some works have reported the performances of SC ethanol for a selective lignin conversion [20,25], the capability of SC ethanol to attack the carbohydrate fraction is rather controversial [26,27]. A priori, these discrepancies might be caused by differences in experimental conditions (batch vs flow reactor, temperature, pressure, and residence time) and by difficulties in analysis such as differentiated measurements of unconverted lignin and chars.
Previously, we have developed an experimental methodology including analytical methods to perform LCB liquefaction in SC ethanol with mass balances greater than 90%. Compositional analysis of the recovered solids by classical acid hydrolysis and analysis based on infrared (Fourier-transform infrared, FTIR) spectroscopy were used to quantify the lignin fraction in the recovered solid residue, which contained both unconverted LCB and chars [28]. The objective of the present work was to investigate the influence of the main experimental parameters on pinewood liquefaction in SC ethanol using this analytical approach. A key point of this work is to get reliable insights on the impact of the each experimental parameters (temperature, time, and density of the SC phase) on the fractionation selectivity at two levels: (1) the transformation of each constituent biopolymer, cellulose, hemicellulose, and lignin, and (2) the product distribution among gases, light products, bio-oils, and chars.
2 Experimental part
2.1 Materials
Feedstocks used in this work are sawdust of pinewood, a solvolysis lignin (provided by CIMV [29]) and a microcrystalline cellulose (purchased from Sigma Aldrich). Wood chips are ground and sieved, and only particles smaller than 500 μm are used. They are lyophilized (soft drying) before use to keep their moisture content constant for all experiments. Ethanol (provided by Sigma Aldrich) has a 99.8% purity and is kept anhydrous by adding molecular sieves. A 72 wt % sulfuric acid (provided by Sigma Aldrich) is used for the acid hydrolysis of solid biomass.
2.2 Protocol of fractionation
The batch type reactor has an internal volume of 74 mL, which can be used at a pressure up to 600 bar and a temperature up to 350 °C. First, 2.5 g of lyophilized pinewood and 27 g (or 34.8 g in a few test reactions) of anhydrous ethanol are introduced in the autoclave. The reactor is closed and cooled down at −59 °C in a dry ice–ethanol mixture. Then, gases are evacuated from the autoclave to reach the final pressure of 100 mbar. The mixture of solvent and biomass generates an autogenous pressure without adding any inert gas. This allows us to reach rigorously the critical coordinates because the volume is set by the apparatus and the initial mass of the solvent is set to lead to a fixed density assuming that the solvent is not significantly consumed during the transformation. The reactor, once warmed up at ambient temperature, is heated up to the reaction temperature (220, 250, or 280 °C) at 5 °C min−1 and kept at this temperature for 0 or 60 min. At the end of the reaction, the reactor is quenched in a cold water bath (0 °C). The recovery protocol of products is described in Scheme 1. The gaseous products are collected in a vessel (95 mL) pre-evacuated at 100 mbar. This vessel is equipped with a manometer to measure the residual pressure. The vessel can be isolated and connected to a gas chromatograph equipped with a thermal conductivity detector for identification of the formed gases. The gas yield is calculated by assuming that light gases contain only one carbon atom with the maximum molecular weight, that is, CO2. The liquid and solid products are separated using a 0.45 μm porosity filter. Then, the unconverted biomass and solid products are washed three times with 30 mL ethanol. The autoclave is also washed with 60 mL acetone to recover potential solids left on the autoclave walls. Solids recovered by filtration are lyophilized for further analysis and designated as “solid residue”. The liquid from reaction medium plus washing ethanol and acetone, designated as “liquid products”, are first analyzed by gas chromatography–mass spectrometry (GC–MS) and then evaporated at 50 °C under reduced pressure, 200 mbar, to eliminate ethanol and acetone. The fraction of light products, previously quantified by GC–MS with a retention time inferior to 20 min [17], are lost during this step. At the end of this evaporation step, a bio-oil fraction is obtained.

Experimental procedure for the product separation/recovery.
The yields of bio-oil and the light products were calculated as follows:
| Yield bio-oil (wt %) = 100 × (weight of bio-oil/weight of dry biomass) |
| Yield light products (wt %) = 100 × (weight of light products/weight of dry biomass) |
2.3 Analytical methods
Acid hydrolysis is used to get the compositional analysis of the solid biomass. In general, cellulose, hemicelluloses, and lignin contents of initial wood can be determined by this method. The acid hydrolysis protocol and monomeric sugar analysis by high-performance liquid chromatography applied in this work are adapted from an National Renewable Energy Laboratory (NREL) method and are described in detail elsewhere [17]. A method was developed to quantify the lignin content in the solid biomass residue based on the integration of the peak area at 1514 cm−1 characteristic of lignin vibration. This method is reported in detail in a previous work [17]. The FTIR spectra were obtained from solid samples diluted in KBr pellet (1%) using a Bruker spectrometer with 2 cm−1 resolution, in absorbance mode. The relative error of the compositional analysis is lower than 5%.
Liquid products (ethanol and acetone solutions) were analyzed by GC–MS using a Shimadzu GC–MS 2010 apparatus equipped with a column NUKol (0.25 μm, 0.25 mm, and 30 m) [17].
Solid-state 13C CP–MAS (cross-polarization–magic-angle sample spinning) nuclear magnetic resonance (NMR) spectra were recorded at a 13C frequency of 100.63 MHz with CP–MAS, using a Bruker DSX 400 NMR spectrometer. Tetramethylsilane was used as an internal standard for the calibration of the chemical shift.
3 Results and discussion
3.1 Pinewood conversion in dense ethanol: influence of the temperature
Three different temperatures, 220, 250, and 280 °C, reached experimentally in less than 1 h with a rate of 5 °C min−1, were studied. Pressures were kept constant during the course of the reaction (Table 1). Experiments were carried out under SC ethanol conditions, except for the run performed at 220 °C (SC ethanol Tc = 241 °C, Pc = 61 bars, and ρc = 0.275 g cm−3). The material balances were calculated as the ratio of all experimental product weights (gases, light products, bio-oils, and solid residue) and initial biomass weight. The “light product” fraction corresponds to the compounds lost during the evaporation step applied to recover the bio-oil fraction. Mass balances superior than 100% were obtained, resulting from the solvent reaction with some products as evidenced by the GC–MS analysis of the liquid part, which shows the formation of esters and ethers in addition to furanic and C6 aromatic derivatives without significant evolution of their relative amounts with the increase in temperature (Supplementary data S1). The yields of gas products were very low (less than 3 wt %) irrespective of the run temperature. These low amounts of gases seem to be specific to biomass liquefaction using SC alcohols in contrast to liquefaction in SC water. Similarly, the yield of light products did not show great changes within the temperature range of 220–250 °C (Table 1). However, at 280 °C, the yield increased by twofold, as compared to product yield at 250 °C, whereas its composition remained unchanged. Besides, the bio-oil yields progressively increased with the temperature from 23 wt % at 220 °C up to 33 wt % at 280 °C.
Liquefaction of pinewood in dense ethanol—influence of the temperature.
| T (°C) | P (bar) | Gas yielda (wt %) | Light product yieldb (wt %) | Bio-oil yield (wt %) | Solid residue (wt %) | Mass balancec (wt %) |
| 220 | 41 | 1 ± 0.05 | 5 ± 0.2 | 23 ± 1.1 | 72 | 101 |
| 250 | 69 | 3 ± 0.1 | 6 ± 0.3 | 27 ± 1.3 | 72 | 108 |
| 280 | 102 | 2 ± 0.1 | 10 ± 0.5 | 33 ± 1.6 | 60 | 105 |
a Assumption made that all produced gases are CO2.
b Light products: products identified and quantified by GC–MS with a retention time inferior to 20 min.
c wt % of all products (light products, gases, bio-oils, and solid residue) on initial solid biomass.
Thus, the total yield of liquid products achieved 43 wt % at 280 °C, whereas the solid residue yield was 60 wt %. The bio-oil yield at 280 °C is relatively close to the highest bio-oils yields reported in the literature from LCB liquefaction in SC alcohols. Indeed, reported bio-oil yields are within 10–50 wt % in SC ethanol [9,14,27]. Bio-oils yields higher than 50 wt % are usually obtained upon addition of catalysts and/or hydrogen [7,13,30].
The compositional analysis of initial pinewood and the solid residues obtained at different temperatures is summarized in Table 2. The results indicate that the lignin content of the solid residue is significantly reduced during the treatment in liquid ethanol at 220 °C, whereas the hemicelluloses content remains almost unchanged. One can note that the subcritical medium (220 °C) or that close to the critical point (250 °C) leads to solid residues of similar composition, which confirms the limited impact of the temperature increase in the range 220–250 °C. In contrast, the solid residue formed after a treatment at 280 °C is strongly changed in composition, with lower proportions of lignin and hemicelluloses and an increase in the char content up to 26 wt %.
Compositional analysis of initial pinewood and solid residues—influence of the temperature.
| Solid | Cellulose (wt %) | Hemicelluloses (wt %) | Lignin (wt %) | Chars (≠lignin) (wt %) |
| Initial pinewood | 37 ± 1.8 | 19 ± 0.9 | 31 ± 1.5 | 0 |
| Solid residue T = 220 °C | 49 ± 2.4 | 17 ± 0.8 | 18 ± 0.9 | 12 ± 0.6 |
| Solid residue T = 250 °C | 49 ± 2.4 | 17 ± 0.8 | 21 ± 1.0 | 10 ± 0.5 |
| Solid residue T = 280 °C | 52 ± 2.6 | 10 ± 0.5 | 8 ± 0.4 | 26 ± 1.3 |
From the compositional analysis of the solid residues and the weight of residual solids, the conversion of each pinewood component was calculated (Fig. 1). The data reported in Fig. 1 underline the peculiar resistance of cellulose toward liquefaction: the cellulose conversion is significant at 280 °C only, with a conversion of 20%. Close to the critical point or in liquid ethanol, at 220 °C, cellulose is not attacked, whereas the conversion of 40% and 50–55% are achieved for hemicellulose and lignin, respectively. At 280 °C, only 15% of the initial lignin remains unconverted against 30% for the hemicellulose part. These data show that under subcritical or SC ethanol treatment, lignin and hemicellulose are very reactive.
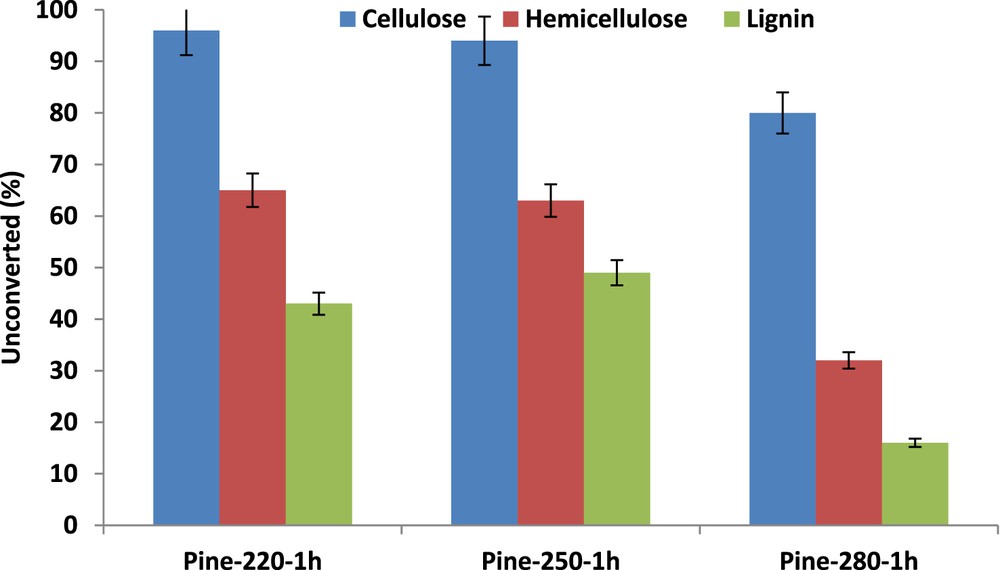
Pinewood liquefaction in dense ethanol at 220, 250, and 280 °C: residual fractions (wt %) of cellulose, hemicelluloses and lignin.
Moreover, lignin appears to be less robust than hemicellulose in SC ethanol. Although the resistance of cellulose in SC ethanol was already reported [31], the reactivity of lignin versus that of hemicellulose remains rather controversial [32]. Biomass conversion and yields in the different fractions (gases, light products, bio-oils, and chars) were determined from the previous experimental data (Fig. 2). It is observed that an increase in temperature from 220 to 250 °C leads to almost equivalent biomass conversion and product yields. In contrast, liquefaction in SC ethanol at 280 °C, as compared to that performed at the critical point, leads to a marked increase in biomass conversion and only a moderate increase in liquid products as explained by the favored production of char at 280 °C.
Pinewood liquefaction in dense ethanol at 220, 250, and 280 °C: pinewood conversion, yields in bio-oils, light products, gases, and char as a function of the temperature.
The solid residues were characterized by 13C MAS-NMR (Supplementary data S2) and FTIR spectroscopic techniques (Fig. 3). The 13C MAS-NMR spectra do not show marked changes with the temperature, except an increase of a broad resonance in the 110–170 ppm range, the typical chemical shifts of aromatic carbons. This is not surprising because the sharp resonance lines are mainly due to cellulose, which was seen to resist to the liquefaction in SC ethanol conditions. The emerging broad resonance lines in the chemical shift range of aromatics support the formation of chars. In other respects, FTIR spectra of the solid residues are more informative of changes in the wood chemical structure (Fig. 3). The band at 1514 cm−1, characteristic of lignin, is strongly reduced as expected from the results reported above. In addition, modifications occur in the νCO vibration range of the acetyl groups. Initially observed at 1740 cm−1, the remaining absorption band in the residue spectrum is observed at lower frequencies, ∼1700–1720 cm−1, which correspond to the wavenumber range of carboxylic groups. This suggests that carboxylic groups might be formed in the solid residue at the expense of acetyl groups in hemicellulose.
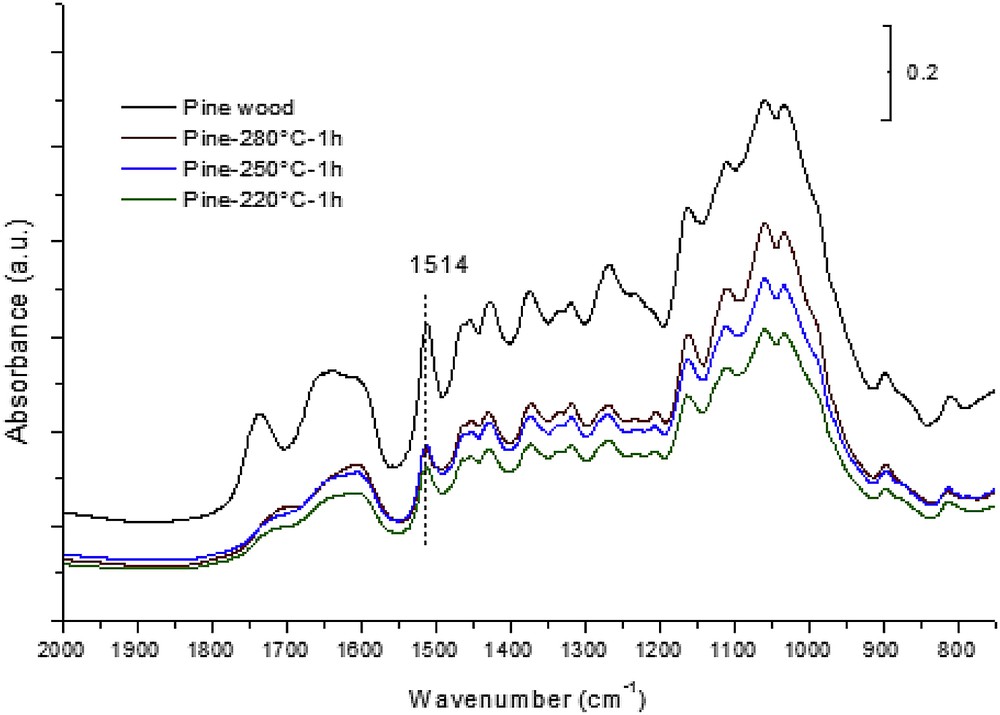
FTIR spectra—solid residues recovered from pinewood liquefaction in dense ethanol at 220, 250, and 280 °C.
3.2 Pinewood liquefaction in dense ethanol: influence of treatment time
Experimental data obtained from increasing treatment times are reported in Table 3. Time zero corresponds to the time when the set temperature was achieved, that is, after less than 1 h. We can observe that the isotherm treatment, performed at 280 °C for 1 h, results mainly in an increase in the yield of liquid products, the formation of gases remaining negligible (<2 wt %), in agreement with the unchanged pressure observed during the treatment. Otherwise, the duration of the treatment at 280 °C has a significant impact on the composition of the solid residue (Table 4). Increased time leads to a pronounced decrease in the lignin content, followed by the less marked decrease in the hemicellulose percentage. This is compensated by an increase in the char content up to 26 wt % after 1 h of treatment, although the cellulose content increased only slightly.
Liquefaction of pinewood in SC ethanol—influence of the treatment time.
| Time (h) | P (bar) | Gas yielda (wt %) | Light product yieldb (wt %) | Bio-oil yield (wt %) | Solid residue (wt %) | Mass balancec (wt %) |
| 0 | 102 | 1 ± 0.05 | 6 ± 0.3 | 27 ± 1.3 | 72 | 106 |
| 1 | 102 | 2 ± 0.1 | 10 ± 0.5 | 33 ± 1.6 | 60 | 105 |
a Assumption made that all produced gases are CO2.
b Light products: products identified and quantified by GC–MS with a retention time inferior to 20 min.
c wt % of all products (light products, gases, bio-oils, and solid residue) on initial solid biomass.
Liquefaction of pinewood in SC ethanol—influence of the treatment time on the solid residue composition.
| Solid | Cellulose (wt %) | Hemicelluloses (wt %) | Lignin (wt %) | Char insoluble (wt %) |
| Fresh pinewood | 37 ± 1.8 | 19 ± 0.9 | 31 ± 1.5 | 0 |
| Solid residue time = 0 h | 48 ± 2.4 | 16 ± 0.8 | 20 ± 1.0 | 10 ± 0.5 |
| Solid residue time = 1 h | 52 ± 2.6 | 10 ± 0.5 | 8 ± 0.4 | 26 ± 1.3 |
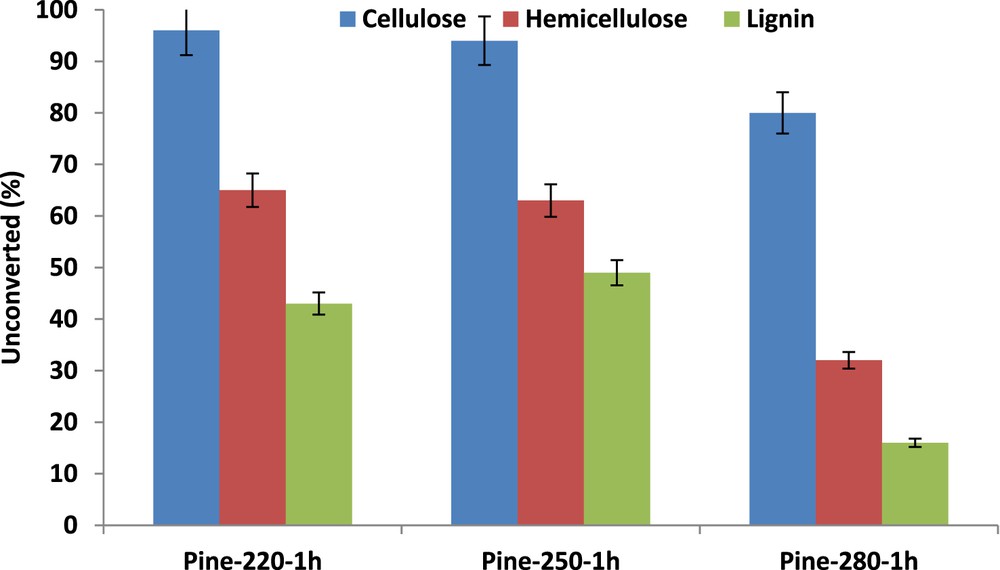
Data reported in Fig. 4 show that about half of the initial lignin and hemicellulose was already liquefied at time zero, whereas the cellulose fraction was not attacked. The treatment time was seen to impact strongly the biomass conversion, with an increase from 35% to 56% when time was extended to 1 h at 280 °C in SC ethanol (Fig. 5). However, the char yield was most strongly favored, increasing up to 15 wt % after 1 h (Fig. 5). Note that this occurs at the expense of the yield of liquid products, which shows only a moderate increase with the severity of the treatment. The impact of the severity of the treatment via enlarged treatment time confirmed the ranking between the different biopolymers and their resistance to their conversion in SC ethanol: biopolymers resistance in SC ethanol: cellulose ≫ hemicellulose > lignin.
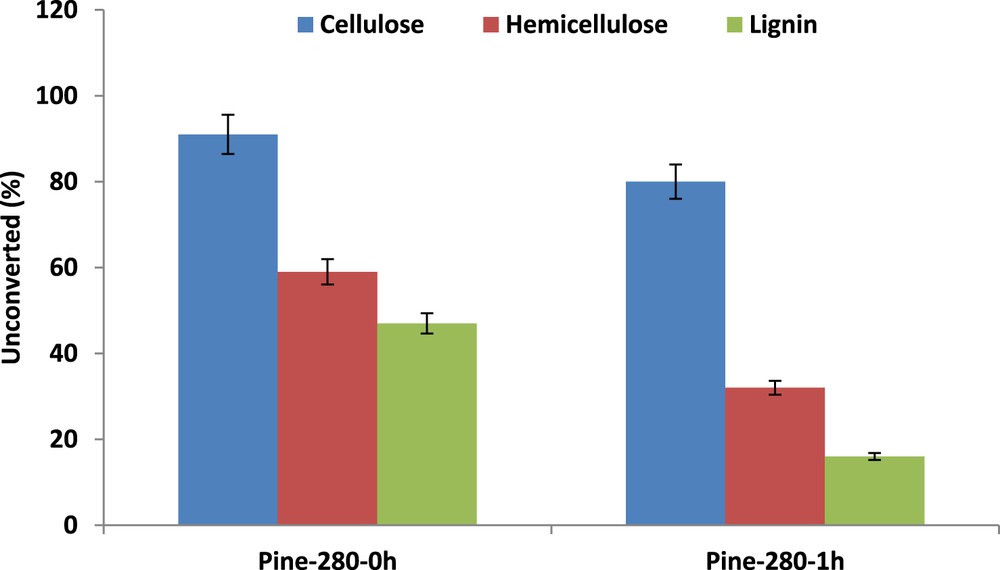
Pinewood liquefaction in SC ethanol at 280 °C, influence of treatment times. Unconverted lignin, cellulose, or hemicellulose in wt %.
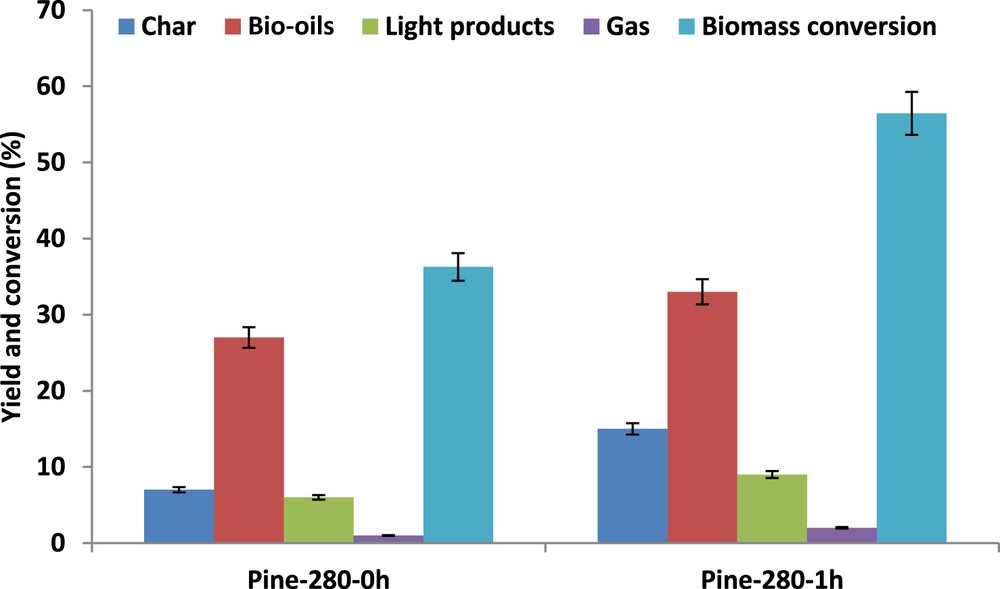
Pinewood liquefaction in SC ethanol at 280 °C, influence of treatment times on pinewood conversion, yields in bio-oils, light products, gas, and char.
One can say that the presence of SC ethanol changes the reactivity of the wood biopolymers. Indeed, under “pure” thermal degradation as that observed in biomass torrefaction, the ranking is: cellulose ≫ lignin > hemicellulose.
The FTIR spectrum of the solid residue mainly confirms the removal of lignin (Supplementary data S3). In contrast, the 13C MAS-NMR data showed the progressive increase in the peak at 147 ppm and the modification in the resonance lines due to aromatic carbons (Supplementary data S4). These modifications occur in line with the significant increase in the char yield with time. The resonance at 147 ppm was previously ascribed to carbon belonging to the aromatic ring of lignin [17]. From the FTIR spectra, which show the decay of the lignin chemical structure, and from the 13C MAS-NMR spectra, which suggest a favored contribution of aromatic rings in the solid residue, one may propose that a repolymerization of liquefied lignin fragments takes place [33].
3.3 Pinewood liquefaction in dense ethanol: influence of SC fluid density
The impact of SC fluid density was scarcely investigated in batch conditions. In the present work, density effect is studied close to the critical point, at 250 °C. This was achieved under isothermal conditions, by feeding the batch autoclave with an increased amount of ethanol. These experiments (Table 5), evidenced the lack of influence on both the solid residue and the light product yields, only yields in bio-oil and gases were slightly increased by an increase in SC fluid density. Also, we can remark that the conversion of the individual biopolymer was not strongly influenced by the SC ethanol fluid density (Supplementary data S5). However, the compositional analysis of the solid residues reveals subtle changes such as the unexpected reduction of char formation with the increase in the SC ethanol density (Table 6). The marked effect of the fluid density is shown in Fig. 6, which evidences the bio-oil yield increase and the parallel decrease in the char yield, from 8 to 3 wt % in dense SC ethanol. Note that yield of the light products remains unchanged. These results show the beneficial impact of the SC ethanol density on the bio-oil formation and on the prevention of char formation. The characterizations of the solid residues by 13C NMR and FTIR as a function of the SC fluid density are shown in S6 and S7 of Supplementary data. If FTIR analysis does not reveal differences, one can observe in the 13C MAS-NMR spectrum of the solid residues obtained in “standard” SC fluid density, the improved intensity of the resonance at 147 ppm, tentatively ascribed to the recombination of lignin fragments. This supports the reduced formation of char in high density SC ethanol.
Liquefaction of pinewood in SC ethanol—influence of the density.
| m solvent (g) | ρ (cm3 g−1) (P bar) | Gas yielda (wt %) | Light product yieldb (wt %) | Bio-oil yield (wt %) | Solid residue (wt %) | Mass balancec (wt %) |
| 27.0 | 0.37 (69 bar) | 0.6 ± 0.03 | 6 ± 0.3 | 27 ± 1.3 | 72 | 106 |
| 34.8 | 0.47 (82 bar) | 1.4 ± 0.1 | 6 ± 0.3 | 32 ± 1.6 | 72 | 111 |
a Assumption made that all produced gases are CO2.
b Light products: products identified and quantified by GC–MS with a retention time inferior to 20 min.
c wt % of all products (light products, gases, bio-oils, and solid residue) on initial solid biomass.
Compositional analysis of the solid residue recovered from pinewood liquefaction in SC ethanol of different densities.
| Solid | Cellulose (wt %) | Hemicellulose (wt %) | Lignin (wt %) | Chars insoluble ≠lignin (wt %) |
| Initial spruce wood | 37 ± 1.8 | 19 ± 0.9 | 31 ± 1.5 | 0 |
| Solid residue ρ = 0.37 g cm−3 | 49 ± 2.4 | 17 ± 0.8 | 21 ± 1.0 | 10 ± 0.5 |
| Solid residue ρ = 0.47 g cm−3 | 49 ± 2.4 | 18 ± 0.9 | 25 ± 1.2 | 4 ± 0.2 |
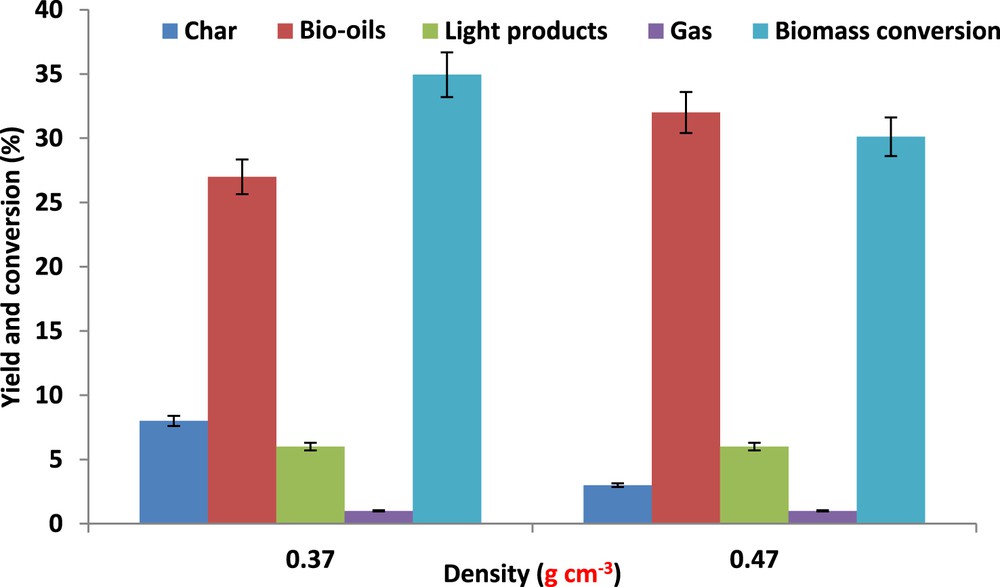
Pinewood liquefaction in SC ethanol at 250 °C, influence of the fluid density on the yields in bio-oils, light products, char, and gases.
These data have underlined the marked positive impact of the SC fluid density (and/or pressure) on the yield in bio-oils and on char formation at 250 °C. The Hildebrand solubility parameter δ of an SC fluid (δ = 0.47Pc1/2ρ) depends on its critical pressure Pc and density ρ. The increase in these solvent properties of SC ethanol fits well with the observed increase in bio-oil yield and the limited formation of chars. Therefore, we can propose that the well-known clustering effect, which occurs in dense SC fluid, might prevent the repolymerization of liquefied high molecular weight constituents of the bio-oil. The absence of the effect of the density on the light product yields may simply indicate that the liquefied light products do not readily form solid residues by recombination and precipitation.
It is noteworthy to observe that the sum of product yields overreached significantly the biomass conversion when the liquefaction test was performed in dense SC ethanol (a liquid like SC fluid) (Fig. 6). Most likely, the insertion of ethyl groups in the products could be an explanation. The favored formation of ethyl products, when dense SC ethanol is used, may be explained by a previously observed phenomenon [34] that using dense SC fluid favors bimolecular reactions. In the present case, reactions between biomass derivatives and ethanol may be favored when a liquid like SC ethanol fluid is used. In contrast, in gas like SC ethanol, recombinations of fragments, issued from LCB liquefaction, could not be limited by the formation of ethanol clusters around LCB fragments, leading to the formation of important amounts of chars.
4 Conclusions
The fractionation of pinewood was studied in subcritical and SC ethanol, using a batch type reactor. The aim was to show the influence of the main experimental parameters (i.e., temperature, time, and SC ethanol density) to get a better control of the wood component fractionation and on the product distribution among light products, bio-oils, gases, and char.
- - Tuning the treatment temperatures from 220 °C (subcritical ethanol) to a value close to the critical temperature, 250 °C, has no impact on the biomass conversion or on the liquid product yields. Under these conditions only lignin and hemicellulose are significantly liquefied with conversion near 40% and 50%, respectively, showing the higher reactivity of lignin as compared to hemicellulose in subcritical and near-critical ethanol. Cellulose was only partially liquefied, 20 wt %, when the temperature reached 280 °C in SC ethanol state. Considering the product selectivity, char formation was the most favored fraction under temperature increase.
- - The treatment time in SC ethanol at 280 °C influences mainly the char formation. 13C MAS-NMR and FTIR spectroscopy analyses of the solid residues suggest that part of the char comes from lignin fragments redeposition.
- - SC ethanol fluid density does not change the conversion of individual constitutive biomass component but has a marked effect on bio-oil yield and prevents char formation. This is tentatively explained by the clustering effect, which prevails in dense SC fluids such as ethanol might prevent the condensation/precipitation of the large molecules, constitutive of bio oils.
A step further will be to investigate the impact of solid catalysts addition, first on each wood component conversion and then on the product distribution and yields.
Acknowledgments
The authors gratefully acknowledge the financial support of the Auvergne Rhône-Alpes region (project CMIRA-COOPERA).