

1 Introduction
Achieving high atom economy in gas phase synthesis of new materials demands precursor molecules with adequate volatility and efficient ligand elimination during thermal decomposition [1–7]. For the deposition of palladium films often homoleptic acetylacetonate derivatives have been reported, although the volatility of these complexes is relatively low. One of the most promising precursors for the deposition of Pd films so far is the heteroleptic cyclopentadienyl allyl palladium, which shows a good volatility but only a limited stability [8–10]. For the deposition of PdS, (allyl) palladium dithiocarbamates have been used as precursors [11,12]. This study describes a similar approach with heteroarylalkenols as NˆO chelating ligands [where NˆO is 3,3,3-trifluoro(pyridin-2-yl)propen-2-ol (H-PyTFP), 3,3,4,4,5,5,5-heptafluoro-1-(pyridin-2-yl)penten-2-ol (H-PyHFP), 3,3,3-trifluoro(1,3-benzoxazol-2-yl)propen-2-ol (H-BOTFP), 3,3,3-trifluoro(1,3-benzthiazol-2-yl)propen-2-ol (H-BTTFP), 3,3,3-trifluoro(dimethyl-1,3-thiazol-2-yl)propen-2-ol (H-DMTTFP), and 3,3,3-trifluoro(dimethyl-1,3-oxazol-2-yl)propen-2-ol (H-DMOTFP)]. They bind the metal center through an enolic oxygen as well as the nitrogen atom of a heterocycle, resulting in stable six-membered metallacycles. We have recently [1–7,13–19] reported on the interplay of the electron-withdrawing perfluoroalkyl units present in the alkenol backbone in combination with the heterocycles that enhances the vapor pressure of the metal complexes and at the same time reduces their moisture sensitivity because of the presence of hydrophobic CF units in the ligand skeleton. The combination of the stabilizing nature of these NˆO chelating ligands and the high reactivity of allyl palladium compounds offers a promising strategy to highly volatile and adequately stable precursors for the deposition of Pd films (Fig. 1).
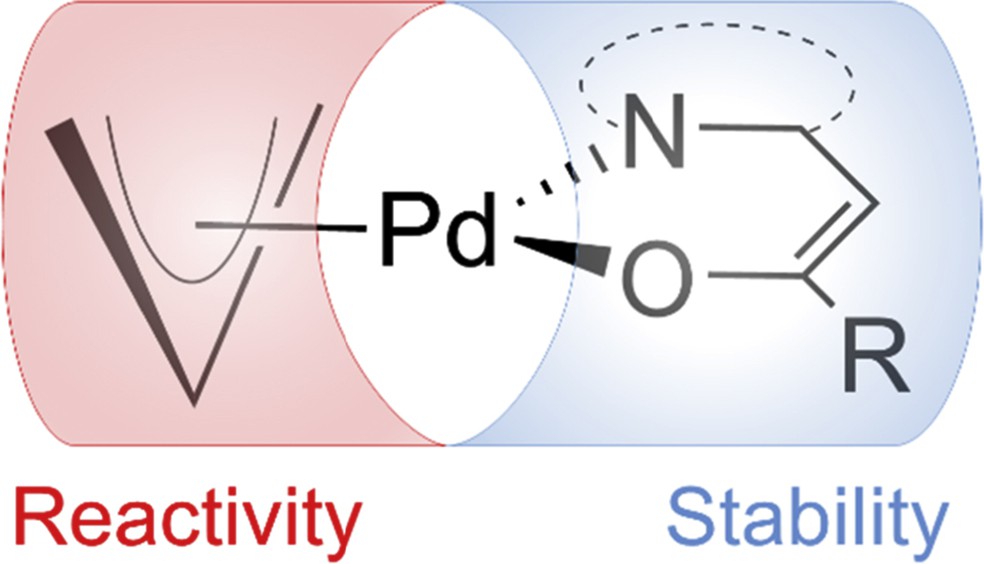
Schematic representation of ligand influence on the stability and reactivity of heteroleptic allyl palladium alkenolates.
2 Results and discussion
2.1 Synthesis and characterization of palladium compounds
By reacting the deprotonated NˆO ligands with bis(η3-allyl)-di-μ-bromopalladium [20], the complexes (NˆO)Pd(η3-C3H5) (Fig. 2) were obtained in high yield and purity. All complexes exist as yellow solids at room temperature and were characterized by multinuclear NMR spectroscopy, EIMS, and by single-crystal X-ray diffraction (XRD) analyses for (PyHFP)Pd(η3-C3H5) (2), and (DMOTFP)Pd(η3-C3H5) (6).
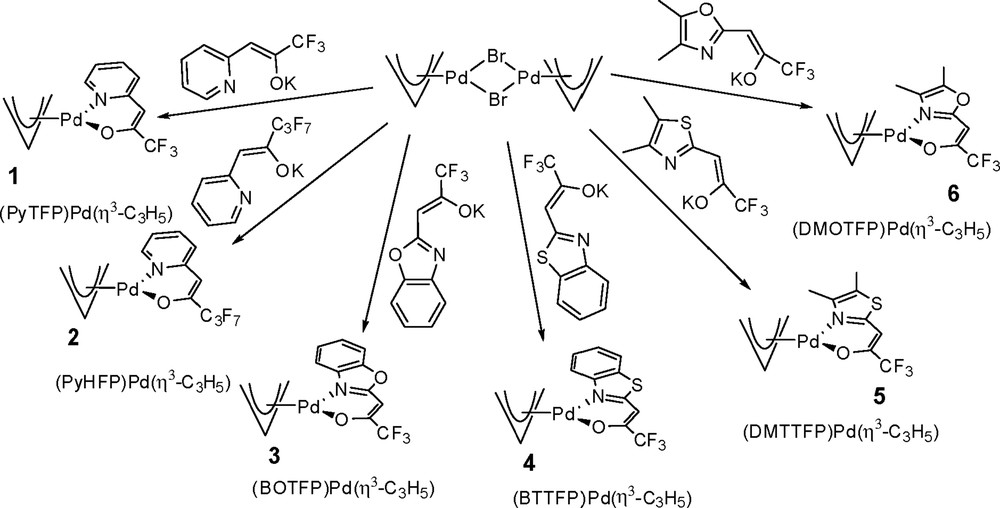
Synthetic approach to the heteroleptic (NˆO)Pd(η3-C3H5) complexes and abbreviations used throughout the article.
Multinuclear NMR data (rt, CDCl3) of complexes 1–6 were recorded by a combination of 13C19F, 13C1H, and 1H1H correlation experiments. The 1H NMR spectra of all six compounds showed one set of signals for the NˆO ligands and an ABCDX spin pattern for the protons of the allyl group because of the asymmetrical nature of the NˆO chelating ligands. The resonances of the protons syn (Hsyn) were observed at a lower field than the anti protons (Hanti) with reference to the central proton. In analogy to other π-allyl complexes, characteristic coupling constants of 3J ∼ 7 Hz for Hanti and 3J ∼ 12 Hz for Hsyn were found in all complexes [21–23]. Inspection of the 1H–1H NOESY spectrum revealed the presence of nuclear Overhauser effect (NOE) contacts, which allowed defining the spatial relationship between the allyl and the protons present in the NˆO ligand unit (Fig. 3).
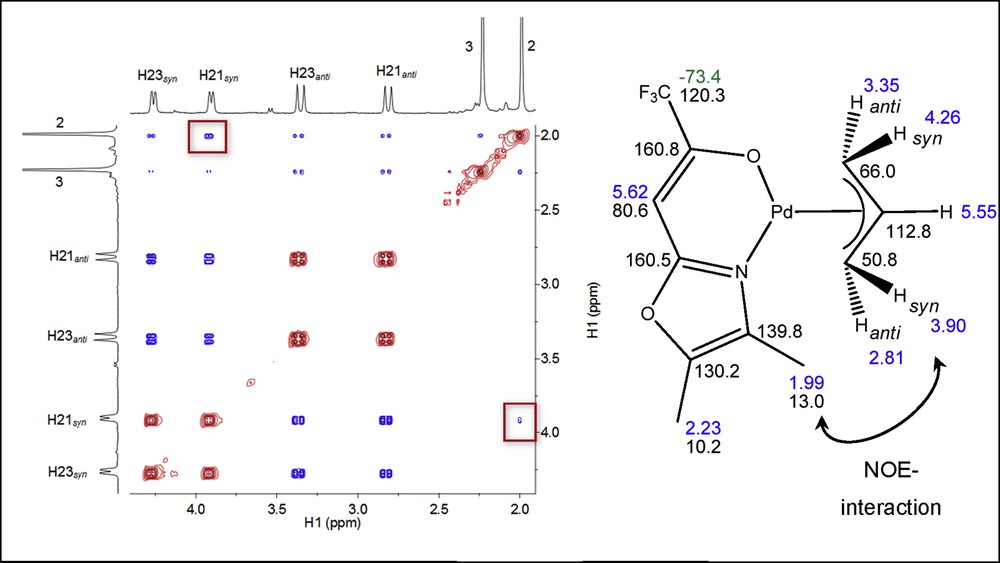
1H–1H NOESY spectrum of complex 6 in CDCl3 at room temperature (left) and structure of 6 with assignment of resonances in ppm (right).
Complex 2 crystallized in the orthorhombic space group Pbca (no. 61, Table 1) whereas 6 crystallized in the monoclinic space group P21/m (no. 11, Table 1) with a mirror plane passing through the whole molecule (Fig. 4). The central palladium atoms in complexes 2 and 6 displayed a slightly distorted square planar arrangement of ligands, with NˆO ligands and one carbon atom of the allyl ligand (C21 and C11) showing an almost ideal planarity. The carbon atoms trans to the nitrogen atoms lie slightly above (0.16(1) Å (C23) and 0.42(2) Å (C9)) the plane, whereas the central carbon atoms lie 0.43(1) Å (C22) and 0.46(1) Å (C10) beneath the plane. The PdO, PdN, and PdC distances are in the range known for other η3-allyl-palladium complexes [24–28].
Selected crystallographic data and collection parameters for compounds 2 and 6.
| Compound | 2 | 6 | ||
| Crystal system | Orthorhombic | Monoclinic | ||
| Space group | Pbca | P21/m | ||
| Cell parameter | a = 7.4935(8) Å | α = 90° | a = 11.149(3) Å | α = 90° |
| b = 19.155 (2) Å | β = 90° | b = 7.380(1) Å | β = 103.32(2)° | |
| c = 20.405 (3) Å | γ = 90° | c = 16.526(4) Å | γ = 90° | |
| Z | 8 | 4 | ||
| Total reflections | 3284 | 5856 | ||
| Unique reflections | 1726 | 2815 | ||
| R1, wR2 [I0> 2σ(I)] | 0.0466, 0.1303 | 0.0336, 0.0615 | ||
| R1, wR2 [all data] | 0.0844, 0.1451 | 0.0800, 0.0718 | ||
| Goodness of fit (GOF) | 0.954 | 0.821 | ||
| CCDC | 1061,689 | 1061,690 |
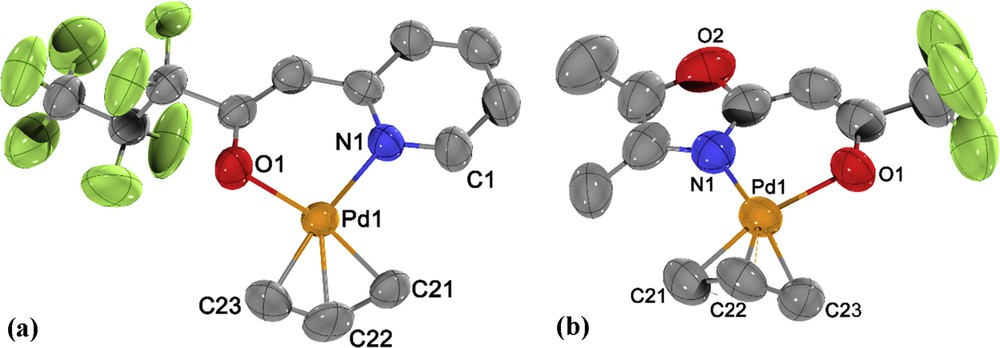
Molecular structure of 2 (a) and 6 (b) with atom labeling scheme. Thermal ellipsoids are drawn at the 50% probability level; all hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): (a) Pd1O1 2.080(4), Pd1N1 2.104(4), Pd1C21 2.119(5), Pd1C22 2.102(6), Pd1C23 2.125(6), O1Pd1C23 97.5(2), O1Pd1N1 91.98(17), C21Pd1N1 102.2(2), C21Pd1C23 68.3(3). (b) Pd1N1 2.087(6), Pd1O1 2.097(5), Pd1C21 2.108(8), Pd1C22 2.080(9), Pd1C23 2.116(9), Pd2N2 2.091(5), Pd2O3 2.091(5), Pd2C24 2.107(8), Pd2C25 2.072(10), Pd2C26 2.130(15); N1Pd1O1 89.8(2), N1Pd1C21 107.6(3), N1Pd1C23 167.0(4), O1Pd1C21 162.6(3), O1Pd1C23 96.3(3), N2Pd2O3 89.4(2), N2Pd2C24 106.7(3), N2Pd2C26 171.4(18), O3Pd2C24 163.9(3), O3Pd2C26 95.7(3).
The thermal stability and volatility of the complexes was found to vary significantly with respect to the coordinated NˆO ligand (Table 2). Complexes (PyTFP)Pd(η3-C3H5) (1), (BOTFP)Pd(η3-C3H5) (3), and (DMOTFP)Pd(η3-C3H5) (6) could be stored under ambient conditions for months without decomposition, whereas (PyHFP)Pd(η3-C3H5) (2) slowly decomposed under reductive elimination to elemental palladium. In contrast, the thiazole derivatives (BTTFP)Pd(η3-C3H5) (4) and (DMTTFP)Pd(η3-C3H5) (5) were only stable for a couple of hours and could not be sublimed without decomposition.
Sublimation temperature and melting points of complexes 1–3 and 6.
| Compound | Melting point | Sublimation temperature |
| 1 | 90 °C | 85 °C/10−3 mbar |
| 2 | 70 °C (decomp.) | 65 °C/10−3 mbar |
| 3 | 135 °C | Decomp. |
| 6 | 90 °C | 65 °C/10−3 mbar |
Considering the volatility of the complexes (Table 2), only 1, 2, and 6 were investigated by thermogravimetric analyses (TGA) (Fig. 5). The TGA profile of compounds 1 and 2 did not show a clear two-step or one-step decomposition, indicating a concomitant loss of allyl and NˆO ligands. Nevertheless, the final residues of 32 wt % in compound 1 and 21 wt % in compound 2 relate to the calculated values for elemental palladium (32 wtcalcd % for compound 1 and 24 wtcalcd % for compound 2) and indicate an efficient conversion of precursor into the targeted solid-state phase. The oxazole-derivative 6 showed a one-step decomposition with a residue of 12 wt %, which is lower than the calculated residue of 30 wtcalcd % implying a partial sublimation of the complex during the measurement.
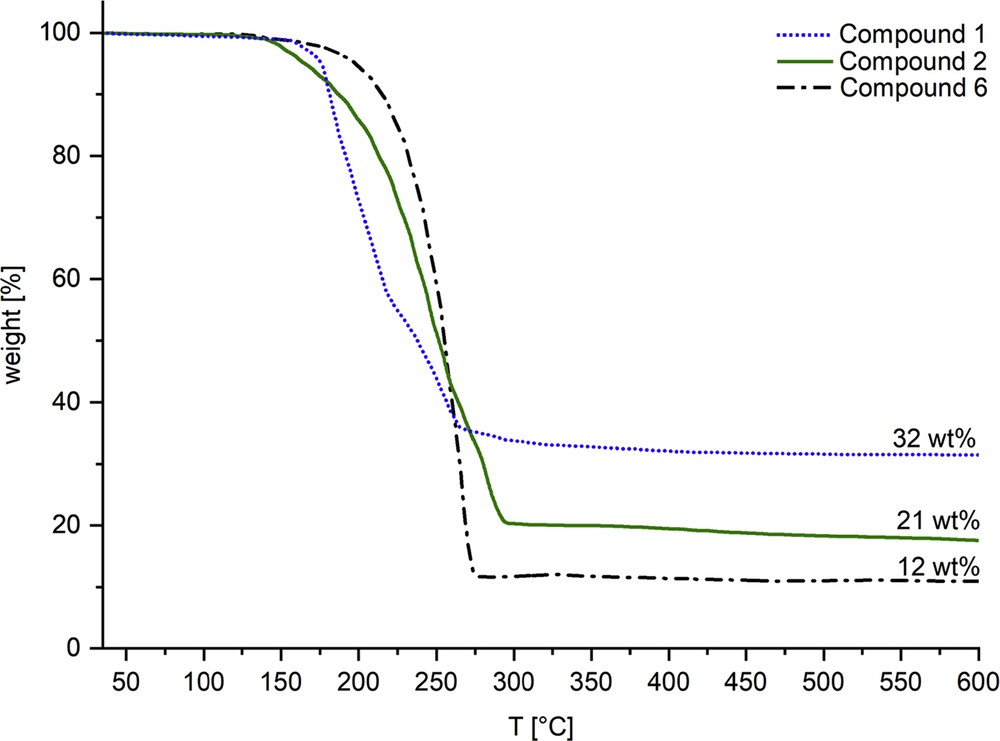
TGA profiles of compounds 1, 2, and 6 measured under nitrogen atmosphere with a heating rate of 10 °C min−1.
Comparison of these results to thermal decompositions (quantitative evaporation during TG/DTA measurements) and sublimation temperatures (90–140 °C/10−3 mbar) of homoleptic Pd(NˆO)2 compounds [14] validated our hypothesis of enhancing the volatility and reducing the stability of the complexes by a judicious choice of ligands. This choice offers stability without hampering the thermal lability, which is essential to initiate the fragmentation cascades enabling the transformation of precursors into materials.
2.2 Plasma-enhanced chemical vapor deposition of Pd films on silicon
Compound 6 was further used for chemical vapor deposition (CVD) studies because of its superior sublimation behavior when compared to compounds 1 and 2. For the generation of palladium films, 6 was decomposed in a plasma-enhanced chemical vapor deposition (PECVD) process using argon as carrier and plasma gas. The XRD patterns showed (Fig. 7a) the as-deposited films on silicon substrates to be completely amorphous. The low deposition temperature supplies an insufficient amount of energy for diffusion and ordering processes resulting in amorphous films. XPS measurements of the same films deposited with changing plasma powers showed in all cases slight contaminations with carbon, oxygen, nitrogen, and fluorine (Fig. 7 of Supplementary Information).
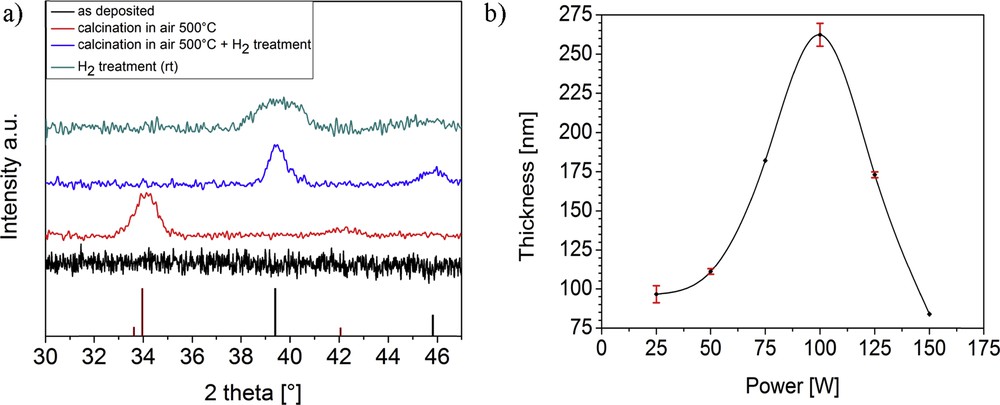
a) Grazing incidence XRD patterns of as-deposited, postannealed, and hydrogen plasma-treated films on Si substrates. All samples were deposited using a plasma power of 75 W (reference patterns: Pd pdf 87-0641 (black), PdO pdf 75-0584 (red)). (b) Film thickness of as-deposited films estimated by cross-sectional SEM depending on the plasma power used for film deposition.
The exemplary high-resolution scan of a sample deposited at a plasma power of 75 W (Fig. 6) confirms the presence of solely palladium(0) in the films; therefore, a complete reduction in the metal ion during plasma processing was achieved. When treating as-deposited amorphous films directly with a hydrogen plasma, the XRD pattern shows an improved crystallinity of the films with broad reflexes that correspond to pure elemental Pd. The broadening of the relatively weak reflexes may be due to remaining carbon contaminations in the coating, which were not removed by the hydrogen plasma treatment.
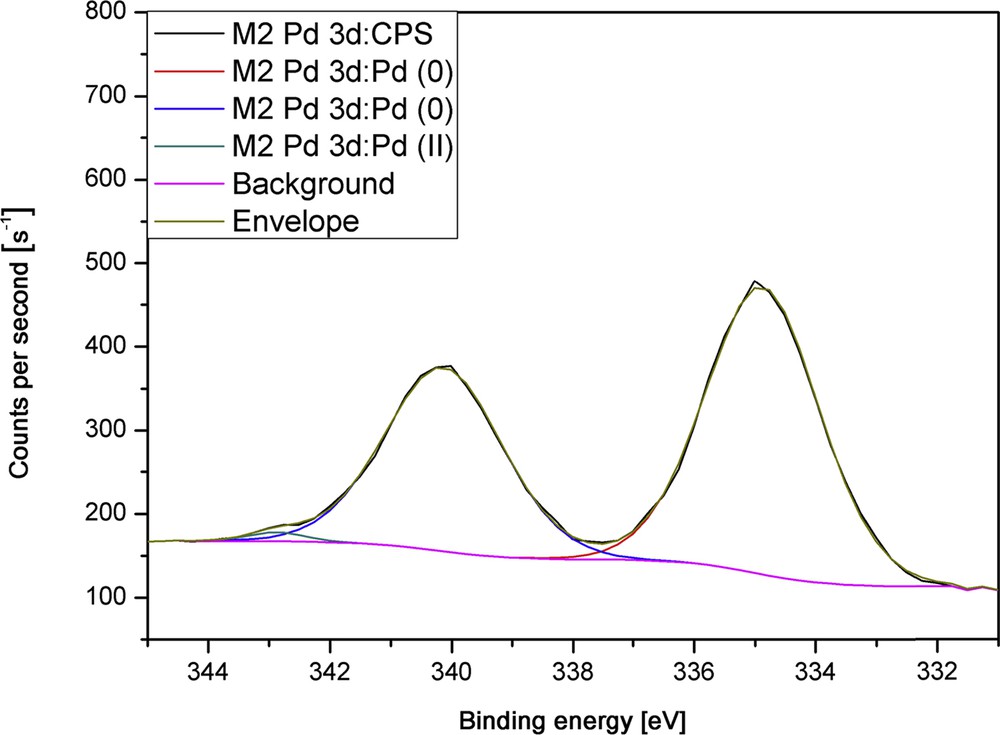
High-resolution XPS measurements of the film deposited at 75 W.
After postannealing the films at 500 °C under air, the XRD pattern exhibits a dominant reflex at 34.1° and two weak reflexes at 42.1° and 54.9°, which correspond to the tetragonal PdO phase. When treating the PdO films with hydrogen plasma, these reflexes disappear and two new reflexes at 39.4 and 45.8° prove the formation of pure elemental Pd, exhibiting relatively sharp signals.
To estimate the influence of used plasma power on film thickness of as-deposited films, cross-sectional SEM images were measured. The thicknesses of coatings depending on the used plasma power are shown in Fig. 7b. With an increasing plasma power, the film thickness increases from 97 nm (25 W) to a maximum of 262 nm (100 W), whereas a further increase in plasma power then leads again to thinner coatings. This trend in film thickness can be explained by the growth-etching competition mechanism [29]. The plasma process is dominated by the decomposition of precursor and therefore by the growth of the film up to a plasma power of 100 W. At a power higher than 100 W, the effect of Ar ion bombardment on the deposited layer dominates over the decomposition of the precursor and leads to an etching of the top surface of the palladium layer instead of a further growth of the layer.
2.3 Solid-supported Pd catalysts on BioC
A favored approach toward CC coupling reactions is based on the use of solid-supported nanostructures or molecular complexes anchored on an inert support such as carbon, zeolites, silicates, biopolymers, or metal oxides (TiO2, Al2O3, etc.) [30–37]. In the scope of sustainability in this study, carbonized wood, which is feedstock derived from renewable resources and has a high surface to volume ratio, has been applied as a support material. As has been shown in previous studies the here used carbonized beech wood (Fagus sylvatica) exhibits a porosity of 59.28 ± 0.20 vol % [38]. The microstructure of the carbon scaffold retains that of the original wood, consisting of interconnected channels of bimodal size distribution (Fig. 8a) with larger pores of 50 μm and smaller pores of 10 μm [39,40]. Because of the high hydrophobicity of the material the often-used wet chemical deposition of Pd particles on the substrates is not applicable. Therefore, CVD was chosen to generate solid-supported catalysts.

a) SEM image of a bare BioC scaffold (b) TEM image of as-deposited Pd NPs on BioC. (c) SEM image of a Pd@BioC catalyst after catalysis reactions (nanoclusters colored in yellow).
On the basis of the investigations of the Pd films on Si substrates, the solid-supported Pd catalysts on carbonized beech wood (Pd@BioC) [41] were generated by PECVD processes with a plasma power of 75 W. The as-deposited Pd nanostructures were then analyzed by TEM, EDX, and ICP-MS measurements to evaluate the morphology and composition of the deposits as well as the amount of palladium on the porous scaffold. As can be seen in Fig. 8b, spherical Pd nanoparticles (NPs) of 8.3 nm (±1.0 nm) are formed during the deposition. The average amount of Pd NPs on the BioC was 0.02 wt %.
As a proof of concept, the Pd@BioC structures were evaluated in established carbon–carbon cross-coupling reactions to determine their catalytic activity and as a comparison commercially available Pd on carbon (Sigma-Aldrich) has been tested. The catalysts were reused in catalytic reactions until no activity could be detected anymore. The Heck reaction is well known to be promoted by Pd NPs as the catalyst reservoir. Therefore, we decided to couple 4-fluoroiodobenzene with n-butyl acrylate in the presence of triethylamine and Pd@BioC (0.091 μmol Pd content; 0.02 wt % Pd) at 100 °C yielding butyl 4-fluorocinnamate in 39% yield (turnover number (TON), 1435) in the first run, which was easily determined by in situ 19F NMR spectroscopy (Fig. 9). Although, in nanocatalysis, it is common to calculate TONs based on surface atoms of NPs following the magic number methodology [42], we decided to determine TONs based on all atoms present, because a change in size and morphology of the particles during the reaction cannot be ruled out. In Fig. 9 the TON of recycling experiments in seven successive runs are depicted. After each run catalyst material was removed from the reaction vessel, washed with a 1:1 mixture of HOiPr and water, and reused with new coupling substrates. In the first five runs the conversion stays relatively stable between 39% and 47% before it drops down to 32% in the sixth and finally to 0% in the seventh cycle. ICP-MS measurements confirmed that this drop of reactivity is on the one hand due to Pd leaching, with only 0.02 μmol Pd left on the substrates after seven catalysis cycles. On the other hand, SEM images of the Pd@BioC after catalysis show nanoclusters that could hamper the reactants to reach remaining catalytically active Pd nanostructures supported on the BioC scaffold (Fig. 8c). Nevertheless, the very low Pd loading of the catalysts gives a total TON of 8767 in all runs. The commercial Pd on carbon only gave a TON of 194. It was not possible to recycle the catalysts at all, which only emphasizes the ease of recyclability of the presented Pd@BioC catalyst.
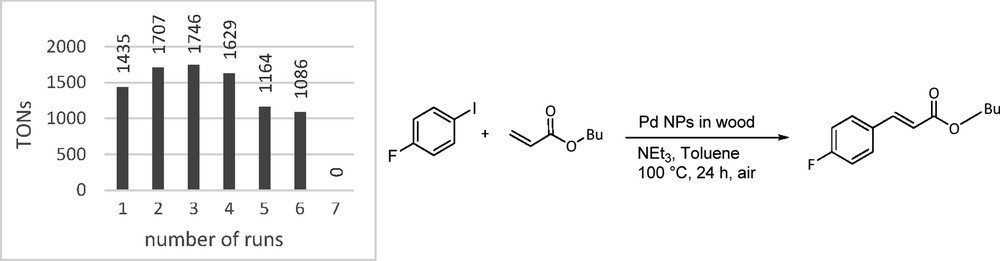
TON of the recycling experiments in the Heck reaction.
Even better results were achieved for the Suzuki coupling of 4-fluoroiodobenzene with phenylboronic acid in water. Potassium carbonate was added as a base and the reaction was run with Pd@BioC (0.091 μmol Pd content; 0.02wt % Pd) at 100 °C yielding 4-fluorobiphenyl in 99% yield (TON, 5229) in the first run (Fig. 10). The catalyst stays stable for the first 15 cycles with yields between 93% and 99% (TONs, 4912–5229). Between cycle 16 and 27 the yield varies from 71% to 99% (TONs, 3750–5229) and then drops down to 52% (TON, 2746) in the 28th run, 11% (TON, 581) in the 29th, and finally no activity in the 30th run. The total TON over 30 cycles reached 139,179, which is remarkably high. As was the case in the Heck reaction, ICP-MS measurements also showed a palladium leaching with only 0.02 μmol Pd left on the substrates after the 30th cycle. It was not possible to determine the palladium leaching of one cycle, because the amount of palladium was under the detection level of the instrument. The commercial Pd on carbon only reaches a TON of 450 and showed, under these reaction conditions, all in all a lower reactivity than the generated Pd@BioC catalyst.
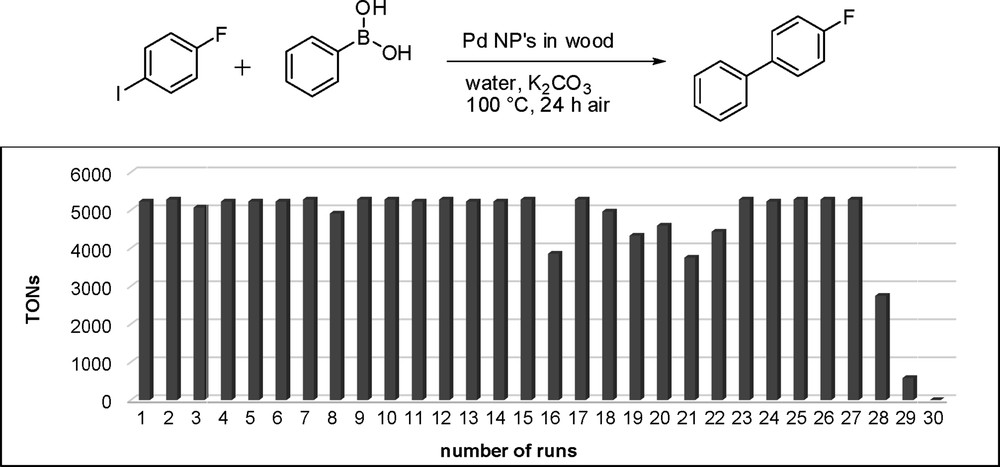
TON of the recycling experiments in the Suzuki reaction.
Both CC cross-coupling reactions demonstrate the potential of the generated solid-supported catalysts. The high surface to volume ratio and bimodal pore distribution of the biomorphic carbon substrates serve well in allowing a deposition of small Pd nanostructures throughout the scaffold. These nanoclusters show a high catalytic activity because of their small size, allowing one to run the Heck and Suzuki reactions with a very low palladium loading. Through the optimization of reaction conditions of the catalytic reactions, along with pretreatment of the support material to enhance the attachment of nanostructured Pd to the wood, even better recyclability of the solid-supported catalysts should be achievable.
3 Experimental section
All manipulations of air and moisture sensitive materials were carried out under nitrogen using Stock-type all glass assemblies. The ligands 3,3,3-trifluoro-1-(pyridin-2-yl)prop-1-en-2-ol (H-PyTFP), 3,3,4,4,5,5,5-heptafluoro-1-(pyridin-2-yl)pent-1-en-2-ol (H-PyHFP), 1-(dimethyl-1,3-oxazol-2-yl)-3,3,3-trifluoropropen-1-ol (H-DMOTFP), 1-(1,3-benzoxazol-2-yl)-3,3,3-trifluoropropen-2-ol (H-BOTFP), 1-(1,3-benzthiazol-2-yl)-3,3,3-trifluoropropen-2-ol (H-BTTFP), and 1-(dimethyl-1,3-thiazol-2-yl)-3,3,3-trifluoropropen-1-ol (H-DMTTFP) were prepared following a procedure described by Kawase et al. [43] As previously reported, the purification process was improved by sublimation of the products instead of column chromatography [1]. Bis(η3-allyl)-di-μ-bromopalladium was prepared according to a published procedure [44]. The wooden templates were prepared by carbonization of wood (beech wood) at 1000 °C in vacuo [41]. The commercial Pd on carbon was bought from Sigma-Aldrich and used without further purification.
Elemental analyses were performed using a HEKAtech CHNS Euro EA 3000. Elemental analyses of the thiazole derivatives (BTTFP)Pd(η3-C3H5) (4) and (DMTTFP)Pd(η3-C3H5) (5) were not possible because of their fast decomposition. Mass spectra were obtained using a Finnigan MAT 95 (20 eV) in m/z (rel%). NMR spectra were recorded using a Bruker Avance II spectrometer operating at 300 MHz, chemical shifts are quoted in parts per million (ppm) relative to TMS (1H and 13C) and CCl3F (19F). XRD patterns were measured using an STOE-STADI MP vertical system working in grazing incident mode, using Cu Kα (λ = 0.15406 nm) radiation. The film thickness and morphology were analyzed by SEM (Nova Nano SEM 430 (FEI)) and TEM (Philips CM300 FEG/UT-STEM). TGA/DTA measurements were performed using a TGA/DSC1 (Mettler-Toledo GmbH, Germany) apparatus. The ICP-MS measurements were carried out by Mikroanalytik Labor Kolbe, Mülheim an der Ruhr, Germany.
Data collection for X-ray structure elucidation was performed using an STOE IPDS I/II diffractometer with graphite monochromated Mo Kα radiation (0.71073 Å). The data were corrected for Lorentz and polarization effects. A numerical absorption correction based on crystal shape optimization was applied for all data. The programs used in this study are STOE's X-Area, including X-RED [45] and X-Shape [46] for data reduction and absorption correction, SIR-92 [47] and SHELXL-97 [48] for structure solution, and SHELXL [48] and ShelXle [49] for structure refinement. The hydrogen atoms were placed in idealized positions and constrained to ride on their parent atom. The last cycles of refinement included atomic positions for all atoms, anisotropic thermal parameters for all non-hydrogen atoms, and isotropic thermal parameters for all hydrogen atoms.
3.1 General synthesis of the organometallic precursor
To a stirred solution of 2 equiv of the NˆO ligand in acetone, 2 equiv of KOH in 1 mL of water was added. This solution was stirred at ambient temperatures for 10 min. In 10 mL CH2Cl2 1 equiv of bis(η3-allyl)-di-μ-bromopalladium was dissolved, added to the reaction mixture, and stirred for 1 h at ambient temperatures. Excess solvent was removed under reduced pressure and the products were purified by sublimation.
3.1.1 (PyTFP)Pd(η3–C3H5) (1)
Yield: 95% (0.18 g). 1H NMR (300 MHz, 298 K, CDCl3) δ: 8.41 (d, J = 5.7 Hz, H1, 1H), 7.57 (d, J = 8.4, 7.3, 1.7 Hz, H3, 1H), 7.08 (d, J = 8.3 Hz, H4, 1H), 6.77 (t, J = 7.2, 5.9, 1.4 Hz, H2, 1H), 5.89–5.77 (m, H22, 1H), 5.63 (s, H6, 1H), 4.08 (dd, J = 7.0, 2.0 Hz, H23syn, 1H), 3.44 (dd, J = 6.8, 2.0 Hz, H21syn, 1H), 3.24 (d, J = 12.4 Hz, H23anti, 1H), 3.08 (d, J = 11.8 Hz, H21anti, 1H). 13C{1H} NMR (75.1 MHz, 298 K, CDCl3) δ: 156.9 (C7), 155.1 (C1), 154.8 (C5), 137.4 (C3), 125.2 (C4), 120.6 (C8), 118.1 (C2), 117.0 (C22), 93.7 (C6), 60.8 (C23), 57.0 (C21). 19F NMR (282 MHz, 298 K, CDCl3) δ: −72.7 (s, 1JC,F = 282 Hz, 2JC,F = 30 Hz). EIMS: 335 (100%, M+), 294 (8%, M+− C3H5), 266 (16%, M+ − CF3), 228 (4%, M+ − C3H5, − CF3), 197 (4%, M+ − COCF3, − C3H5), 147 (22%, M+ − PyTFP). Elemental Anal. Calcd for C11H10F3NOPd: C, 39.37; H, 3.00; N, 4.17. Found: C, 39.62; H, 3.87; N, 4.47. Mp: 90 °C. Sublimation temperature: 85 °C/10−3 mbar.
3.1.2 (PyHFP)Pd(η3–C3H5) (2)
Yield: 87% (0.21 g). 1H NMR (300 MHz, 298 K, CDCl3) δ: 8.40 (d, J = 4.9 Hz, H1, 1H), 7.57 (t, J = 8.3 Hz, H3, 1H), 7.08 (d, J = 8.3 Hz, H4, 1H), 6.77 (t, J = 7.2 Hz, H2, 1H), 5.87–5.74 (m, H22, 1H), 5.62 (s, H6, 1H), 4.00 (dd, J = 7.0, 2.0 Hz, H23syn, 1H), 3.42 (dd, J = 6.8, 1.9 Hz, H21syn, 1H), 3.20 (d, J = 12.5 Hz, H23anti, 1H), 3.04 (d, J = 11.8 Hz, H21anti, 1H). 13C{1H} NMR (75.1 MHz, 298 K, CDCl3) δ: 157.3 (C7), 154.9 (C1), 154.9 (C5), 137.0 (C3), 124.9 (C4), 118.0 (C10), 117.8 (C2), 116.7 (C22), 111.7 (C8), 109.0 (C9), 95.2 (C6), 60.6 (C23), 56.4 (C21). 19F NMR (282 MHz, 298 K, CDCl3) δ: −126.6 (s, 1JC,F = 266 Hz, 2JC,F = 32 Hz, F9, 2F),−117.2 (q, 4JF,F = 9 Hz, F8, 2F),−80.5 (t, 4JF,F = 9 Hz, F10, 3F). EIMS: 435 (80%, M+), 289 (40%, M+ − PyHFP), 39 (8%, C3H4+). Elemental Anal. Calcd for PdC13H10F7NO: C, 35.84; H, 2.31; N, 3.22. Found: C, 35.89; H, 2.70; N, 3.46. Sublimation temperature: 65 °C/10−3 mbar.
3.1.3 (BOTFP)Pd(η3–C3H5) (3)
Yield: 80% (0.18 g). 1H NMR (300 MHz, 298 K, CDCl3) δ: 7.46–7.41 (m, H5, 1H), 7.33–7.27 (m, H2/H3, 2H), 7.27–7.23 (m, H4, 1H), 5.81 (s, H8, 1H), 5.71–5.59 (m, H22, 1H), 4.38 (dd, J = 6.9, 1.7 Hz, H23syn, 1H), 4.23 (dd, J = 6.7, 1.7 Hz, H21syn, 1H), 3.48 (d, J = 12.5 Hz, H23anti, 1H), 2.99 (d, J = 11.6 Hz, H21anti, 1H). 13C{1H} NMR (75.1 MHz, 298 K, CDCl3) δ: 165.4 (C9), 164.8 (C7), 148.2 (C6), 141.2 (C1), 124.5 (C3), 123.4 (C4), 119.7 (C10), 116.5 (C2), 113.0 (C22), 110.5 (C5), 79.9 (C8), 66.2 (C23), 49.6 (C21). 19F NMR (282 MHz, 298 K, CDCl3) δ: −73.8 (s, 1JC,F = 283 Hz, 2JC,F = 33 Hz). EIMS: 375 (100%, M+), 335 (12%, M+ − C3H5), 306 (8%, M+ − CF3), 229 (8%, BOTFP+), 147 (20%, M+ − BOTFP). Mp: 135 °C decomposition. Elemental Anal. Calcd for PdC13H10F3NO2: C, 41.57; H, 2.68; N, 3.73. Found: C, 40.72; H, 2.87; N, 3.49. Sublimation temperature: decomposition.
3.1.4 (BTTFP)Pd(η3–C3H5) (4)
Yield: 85% (0.20 g). 1H NMR (300 MHz, 298 K, CDCl3) δ: 7.69 (d, J = 7.9 Hz, H5, 1H), 7.65 (d, J = 8.0 Hz, H2, 1H), 7.42 (t, J = 8.4 Hz, H3, 1H), 7.30 (t, J = 8.0 Hz, H4, 1H), 5.94 (s, H8, 1H), 5.69–5.57 (m, H22, 1H), 4.30 (d, J = 6.5 Hz, H23syn, 1H), 4.06 (d, J = 6.1 Hz, H21syn, 1H), 3.49 (d, J = 12.4 Hz, H23anti, 1H), 3.03 (d, J = 11.7 Hz, H21anti, 1H). 13C{1H} NMR (75.1 MHz, 298 K, CDCl3) δ: 167.9 (C7), 160.4 (C9), 153.0 (C1), 130.2 (C6), 126.3 (C3), 124.3 (C4), 121.1 (C5), 120.6 (C2), 119.8 (C10), 113.0 (C22), 88.3 (C8), 66.3 (C23), 51.9 (C21). 19F NMR (282 MHz, 298 K, CDCl3) δ: −73.4 (s, 1JC,F = 283 Hz, 2JC,F = 32 Hz). EIMS: 391 (100%, M+), 355 (12%, M+ − C3H5), 322 (8%, M+ − CF3), 245 (40%, BTTFP+), 176 (BTTFP+ − CF3), 147 (24%, M+ − BTTFP). Mp: 87 °C decomposition. Sublimation temperature: decomposition.
3.1.5 (DMTTFP)Pd(η3–C3H5) (5)
Yield: 73% (0.27 g). 1H NMR (300 MHz, CDCl3) δ: 5.81 (s, H4, 1H), 5.63–5.51 (m, H22, 1H), 4.21 (d, J = 6.9 Hz, H23syn, 1H), 3.79 (d, J = 6.6 Hz, H21syn, 1H), 3.38 (d, J = 12.4 Hz, H23anti, 1H), 2.83 (d, J = 11.9 Hz, H21anti, 1H), 2.29 (s, 2-CH3, 3H), 2.22 (s, 1-CH3, 3H). 13C{1H} NMR (75,1 MHz, 298 K, CDCl3) δ: 164.1 (C3), 156.1 (C5), 147.2 (C2), 120.6 (C6), 120.2 (C1), 112.7 (C22), 87.9 (C4), 65.4 (C23), 52.3 (C21), 17.7 (1-CH3), 12.3 (2-CH3). 19F NMR (282 MHz, 298 K, CDCl3) δ: −73.0 (s, 1JC,F = 282 Hz, 2JC,F = 31 Hz).
3.1.6 (DMOTFP)Pd(η3–C3H5) (6)
Yield: 90% (0.18 g). 1H NMR (300 MHz, 298 K, CDCl3) δ: 5.62 (s, H4, 1H), 5.62–5.49 (m, H22, 1H), 4.26 (dd, J = 6.9, 2.2 Hz, H23syn, 1H), 3.90 (dd, J = 6.6, 1.6 Hz, H21syn, 1H), 3.35 (d, J = 12.5 Hz, H23anti, 1H), 2.81 (d, J = 11.9 Hz, H21anti, 1H), 2.23 (s, 2-CH3, 3H), 1.99 (s, 1-CH3, 3H). 13C{1H} NMR (75.1 MHz, 298 K, CDCl3) δ: 160.8 (C5), 160.5 (C3), 139.8 (C1), 130.2 (C2), 120.3 (C6), 112.8 (C22), 80.6 (C4) 66.0 (C23), 50.8 (C21), 13.0 (1-CH3), 10.2 (2-CH3). 19F NMR (282 MHz, 298 K, CDCl3) δ: −73.4 (s, 1JC,F = 282 Hz, 2JC,F = 32 Hz). EIMS: 353 (100%, M+), 313 (24%, M+ − C3H5), 207 (20%, DMOTFP+), 138 (16%, DMOTFP+ − CF3), 39 (8%, C3H4+). Elemental Anal. Calcd for C11H10F3NO2Pd: C, 39.37; H, 3.00; N, 4.17. Found: C, 39.62; H, 3.87; N, 4.47. Mp: 90 °C. Sublimation temperature: 65 °C/10−3 mbar.
3.2 General procedure of the PECVD process
A weighed amount of precursor (100 mg) was transferred into a stainless-steel precursor tank, which was connected to the plasma reactor (plasma electronic) and heated to a temperature of 100 °C to provide a sufficient precursor flow. Argon was used as carrier gas. All substrates were cleaned by ultrasonification in distilled water and isopropanol and by Ar sputtering using 20 sccm Ar and a plasma power of 50 W for 3 min. Silicon and carbon substrates, which were held at room temperature during the deposition, were placed in the reactor near the precursor inlet and the chamber was pumped down to a base pressure of ca. 0.5 Pa before the precursor/argon mixture and an additional amount of argon (20 sccm) as reactive gas were introduced and the processes at variable plasma powers (in a range of 25–150 W) were started. The reaction time for each process was held constant at 1 h.
3.3 CC coupling reactions
3.3.1 Heck reaction
In a typical experiment 4-fluoroiodobenzene (40.7 μL, 0.3 mmol), n-butyl acrylate (42.6 μL, 0.3 mmol), triethylamine (47.3 μL, 0.36 mmol), Pd@BioC (48.5 mg; 0.020 wt % Pd), and toluene (1 mL) were introduced into a 20 mL reaction vial in air. The mixture was stirred at 100 °C for 24 h. The conversion was directly determined by 19F NMR spectroscopy in CDCl3.
3.3.2 Suzuki reaction
In a typical experiment 4-fluoroiodobenzene (43.3 μL, 0.38 mmol), phenylboronic acid (68 mg, 0.56 mmol), potassium carbonate (105 mg, 0.75 mmol), Pd@BioC (37.8 mg; 0.020 wt % Pd), and water (3 mL) were introduced into a 20 mL reaction vial in air. The mixture was stirred at 100 °C for 24 h. After cooling to room temperature, the product was extracted with pentane (30 mL), the organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure. The white solid was analyzed, and the conversion was determined by 19F NMR spectroscopy in CDCl3.
4 Conclusions
Synthesis and characterization of new heteroleptic allyl palladium complexes showed the utility of heteroarylalkenolate ligands in delivering volatile and thermally labile complexes. The thermal stability of the complexes was found to be highly dependent on the heterocycles of the ligand with the thiazole derivatives showing the highest tendency to decompose to Pd(0) under ambient conditions. The most stable complex with a high volatility of 65 °C/10−3 mbar was used as a precursor for the formation of palladium nanostructures in a PECVD process on both flat (Si) and mesoporous (BioC) surfaces.
Palladium nanostructures embedded in porous BioC scaffold showed high catalytic activity and recyclability for Heck and Suzuki reactions, which is further emphasized by the lower TON of commercially available Pd on carbon. In combination with the total TON of 8767 (Heck) and 139,179 (Suzuki) under nonoptimized reaction conditions and an excellent selectivity the potential of the presented sustainable solid-supported catalysts could be demonstrated.
Acknowledgments
The authors gratefully acknowledge the University of Cologne and the Regional Research Cluster Sustainable Chemical Synthesis for financial support. The project “Sustainable Chemical Synthesis (SusChemSys)” is cofinanced by the European Regional Development Fund (ERDF) and the state of North Rhine-Westphalia, Germany, under the Operational Program “Regional Competitiveness and Employment” 2007–2013. Thanks are also due to A. Gutiérrez-Pardo, J. Ramirez-Rico, and J.M. Fernández (University of Sevilla) for providing the BioC substrates.