

1 Introduction
L'exploitation des glutaconates de dialkyle en synthèse a fait l'objet d'un intérêt croissant au cours de ces dernières années. En effet, en raison de leur capacité naturelle à générer facilement des liaisons multiples par l'intermédiaire de processus nucléophiles comme électrophiles, ils ont été utilisés dans une variété importante de réactions domino [1] conduisant à des hétérocycles comme, par exemple, les dérivés de l'isoquinolinedione [2], les dibenzopyranones [3], les phénanthridinones [4], les isoxazolidines [5], des pyrrolidines 2,3,4-trisubstituées [6] ou des arènes densément substitués [7]. Bien que ces travaux proposent de multiples approches à une gamme significative de structures cycliques et hétérocycliques, la synthèse de δ-valérolactones substituées à partir de glutaconate de diméthyle et d'aldéhydes aromatiques n'a pas été décrite à ce jour [8].
Dans des travaux précédents, nous avons décrit l'utilisation de composés benzyl-et arylmétalliques comme nucléophiles dans les réactions à trois et quatre composants [9]. Bien que leur caractère nucléophile soit probablement la caractéristique la plus commune et évidente de la plupart des espèces organométalliques, il convient de garder à l'esprit que ces composés possèdent généralement une basicité significative. Au cours de ce travail, nous avons exploité la basicité du bromure de phénylzinc dans une pseudo-réaction à trois composants entre le glutaconate de diméthyle et des aldéhydes aromatiques. Nous montrons que cette stratégie permet la génération facile de dihydropyranones 2,3,5-trisubstituées selon un processus domino original aldolisation/aldolisation/élimination/lactonisation.
2 Résultats et discussion
Au cours d'un projet consacré à la mise au point de réactions tandem addition conjuguée/aldolisation cobalto-catalysées entre des organozinciques aromatiques, des accepteurs de Michael et des aldéhydes aromatiques, nous avons fait la découverte inattendue de ce que le (E)-5-benzylidène-6-oxo-2-phényl-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3a) pouvait être obtenu avec un bon rendement en mélangeant du glutaconate de diméthyle (2), du benzaldéhyde (la) et du bromure de phénylzinc en excès dans de l'acétonitrile à température ambiante pendant une nuit (Schéma 1).

Le système réactionnel menant à la lactone 3a.
Ce résultat peut être envisagé comme la conséquence de la réactivité limitée des organozinciques aromatiques vis-à-vis des aldéhydes, et surtout de l'acidité importante prévisible en position α du carboxylate de méthyle, en raison de la stabilisation exacerbée de l'espèce déprotonée du fait de la présence des deux fonctions ester en positions α et γ. La détermination précise de la structure moléculaire de 3a a nécessité l'utilisation conjointe de diverses méthodes spectroscopiques. En effet, bien que les expériences de RMN aient déjà fourni des indications significatives concernant la nature cyclique de 3a, la détermination de la stéréochimie E de la partie benzylidène a été effectuée sans ambiguïté à l'aide d'une expérience de diffraction des rayons X (Fig. 1).
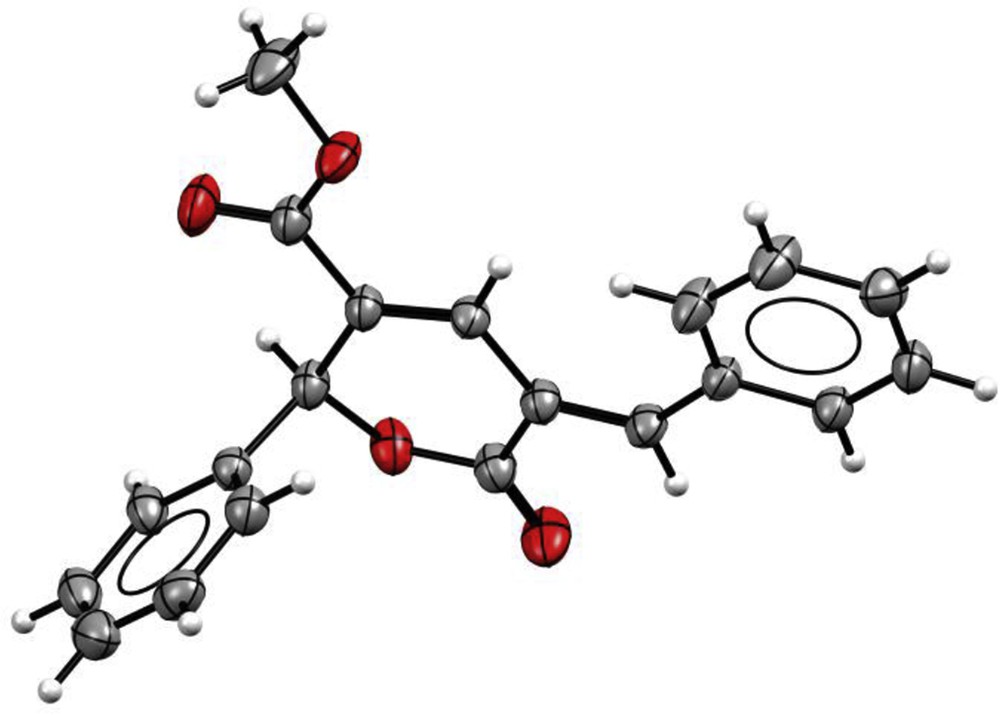
Structure tridimensionnelle de 3a obtenue par diffraction des rayons X [10].
Afin de mieux comprendre le processus menant à la formation de cette lactone à six chaînons, des expériences complémentaires ont été réalisées. Ainsi, des essais impliquant des arylzinciques fonctionnalisés ont indiqué que leur fragment aromatique n'est jamais incorporé dans la structure finale. Afin de confirmer le fait que les deux motifs aromatiques présents sur la structure finale proviennent bien de l'aldéhyde, un essai de réaction utilisant deux équivalents d'un benzaldéhyde fonctionnalisé a été réalisé. Celui-ci montre que deux motifs aromatiques fonctionnalisés sont présents dans la structure finale. Ceci plaide évidemment en faveur d'une simple réaction acide–base induite par l'espèce arylzincique (voir le mécanisme réactionnel proposé plus bas). On peut noter que les organozinciques ne sont pas les seuls nucléophiles carbonés présentant un tel comportement. En effet, Overman et al. indiquent que les organocuprates présentent vraisemblablement un caractère alcalin lorsqu'ils sont utilisés en présence de glutaconates de dialkyle. Néanmoins, leur caractère nucléophile prédominant peut être restauré par l'adjonction de chlorure de triméthylsilyle au milieu réactionnel [11].
Au démarrage de l’étude, nous avons tenté de déterminer si l'utilisation d'un organozincique en tant que base était obligatoire ou non. Par conséquent, plusieurs expériences impliquant diverses bases usuelles et divers solvants ont été réalisées. Les résultats sont indiqués dans le Tableau 1.
Evaluation des conditions réactionnellesa.
| Entrée | Base | Quantité (équiv.) | Solvant | Durée (h) | Rdt (%)b |
| 1 | Pyrrolidine | 2 | CH3CN | 3 | – |
| 2 | NaOH | 2 | CH3CN | 3 | <10 |
| 3 | tBuONa | 2 | CH3CN | 1 | – |
| 4 | tBuOK | 2 | CH3CN | 2 | <5 |
| 5 | NaH | 2 | CH3CN | 1 | <5c |
| 6 | PhZnBr | 2 | CH3CN | 15 | 95d |
| 7 | PhZnBr | 2 | CH3CN | 15 | 61d,e |
| 8 | tBuOK | 2 | MeOH | 1 | <5 |
| 9 | tBuOK | 0.1 | MeOH | 18 | <5 |
| 10 | MeONa | 2 | MeOH | 2 | <5 |
a Les réactions ont été effectuées avec 20 mL de solvant, 2 mL (14 mmol) de glutaconate de diméthyle (2), 0,75 mL (7,5 mmol) de benzaldéhyde (1a) et la base à température ambiante.
b Rendement obtenu par CPG.
c Réaction violente.
d Réaction effectuée avec 40 mL d'acétonitrile, 3 mL (21 mmol) de glutaconate de diméthyle, 1,5 mL (15 mmol) de benzaldéhyde et ∼20 mmol de bromure de phénylzinc.
e Réaction effectuée avec 1,4 mL (10 mmol) de 2.
Comme il ressort clairement de ces résultats, seul le bromure de phénylzinc favorise efficacement la réaction. Des traces du composé désiré ont généralement été observées en utilisant d'autres bases, mais 3a n'a jamais été le produit principal et, en outre, des mélanges complexes ont toujours été obtenus. À la suite de cette étude, nous avons tenté de généraliser la réaction en utilisant divers aldéhydes aromatiques et hétéroaromatiques. Les résultats sont indiqués dans le Tableau 2.
Portée de la réactiona.
| Entrée | Ar = | Produit | Rdt (%)b |
| 1 | Phényl | 3a | 75 |
| 2 | 2-Me-Phényl | 3b | 44 |
| 3 | 4-Me-Phényl | 3c | 35 |
| 4 | 2-MeO-Phényl | 3d | 42 |
| 5 | 3-MeO-Phényl | 3e | 80 |
| 6 | 4-MeO-Phényl | 3f | 40 |
| 7 | 4-Me2N-Phényl | C4 | –c |
| 8 | 2-Furyl | 3g | 40 |
| 9 | 2-Thiényl | 3h | 36 |
| 10 | 3-F-Phényl | 3i | 75 |
| 11 | 4-F-Phényl | 3j | 76 |
| 12 | 3-Cl-Phényl | 3k | 55 |
| 13 | 4-Br-Phényl | 3l | 38 |
| 14 | 2-F3C-Phényl | 3m | 66 |
| 15 | 4-NC-Phényl | – | –d |
| 16 | 4-MeO2C-Phényl | – | –d |
a Les réactions ont été effectuées avec 40 mL d'acétonitrile, 3 mL (21 mmol) de glutaconate de diméthyle (2), 15 mmol d'aldéhyde aromatique 1 et ∼20 mmol de bromure de phénylzinc, à température ambiante pendant une nuit.
b Rendement isolé.
c Un composé nouveau C4 de couleur orange est obtenu avec 53% de rendement (voir plus bas).
d Pas ou peu de lactone formée.
La réaction apparaît possible avec une gamme relativement importante d'aldéhydes aromatiques. Des benzaldéhydes substitués par des groupements électrodonneurs (Tableau 2, entrées 2–6) fonctionnent dans le processus, de même que des aldéhydes halogénés (Tableau 2, entrées 10–13). Un benzaldéhyde substitué par un groupement trifluorométhyle mène à la lactone correspondante (Tableau 2, entrée 14). En revanche, les benzaldéhydes substitués par des groupements à effet mésomère attracteur ne semblent pas permettre la réaction (Tableau 2, entrées 15 et 16). Les essais réalisés avec des aldéhydes hétéroaromatiques (Tableau 2, entrées 8 et 9) montrent qu'ils permettent la réaction, bien que les rendements soient plus faibles dans ce cas. Il semble que les effets stériques soient limités, car les aldéhydes ortho-substitués fonctionnent correctement dans la réaction (Tableau 2, entrées 2, 4 et 14). En revanche, il semble que la présence de groupements à effet mésomère donneur en position ortho ou para limite le rendement réactionnel, comme l'attestent les résultats obtenus avec les substituants OMe en position 2 ou 4 (Tableau 2, entrées 4 et 6). Il est également intéressant de noter que la durée de la réaction peut avoir une importance non négligeable. En effet, même si le produit de couplage est très rapidement détecté dans le mélange réactionnel, la durée optimale est généralement comprise entre 12 h et 18 h, des temps de réaction prolongés ayant parfois mené à une dégradation partielle des composes formés. Le résultat obtenu avec le para-diméthylaminobenzaldéhyde (Tableau 2, entrée 7) constitue une exception, car la lactone attendue n'est pas du tout formée et un composé fortement conjugué C4 est obtenu à sa place [12]. La formation de ce composé de couleur orange peut être expliquée sur la base du mécanisme réactionnel proposé plus bas. Enfin, des essais complémentaires réalisés avec des aldéhydes aliphatiques n'ont pas permis d'obtenir les lactones attendues. De même, l'extension du procédé à une imine activée n'a malheureusement pas mené à la formation du lactame correspondant (Schéma 2).
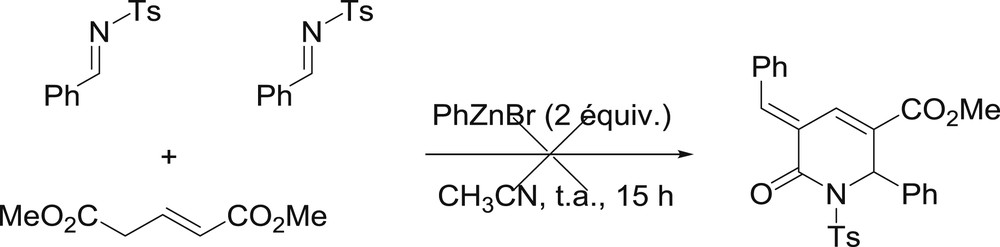
Essai de formation d'un lactame à six chaînons.
Un essai de réaction engageant deux aldéhydes de réactivité présumée différente, le benzaldéhyde et le 2-trifluorométhylbenzaldéhyde, a ensuite été réalisé. Nous supposions qu'il était possible d'observer une sélectivité lors de la première addition sur l'aldéhyde et obtenir ainsi un produit nettement majoritaire. Malheureusement, la sélectivité s'est révélée absente car, quel que soit l'ordre d'introduction des aldéhydes dans le milieu réactionnel, une répartition quasi statistique de ∼25/25/25/25 est obtenue pour les quatre produits formés après 15 h à température ambiante (Schéma 3).
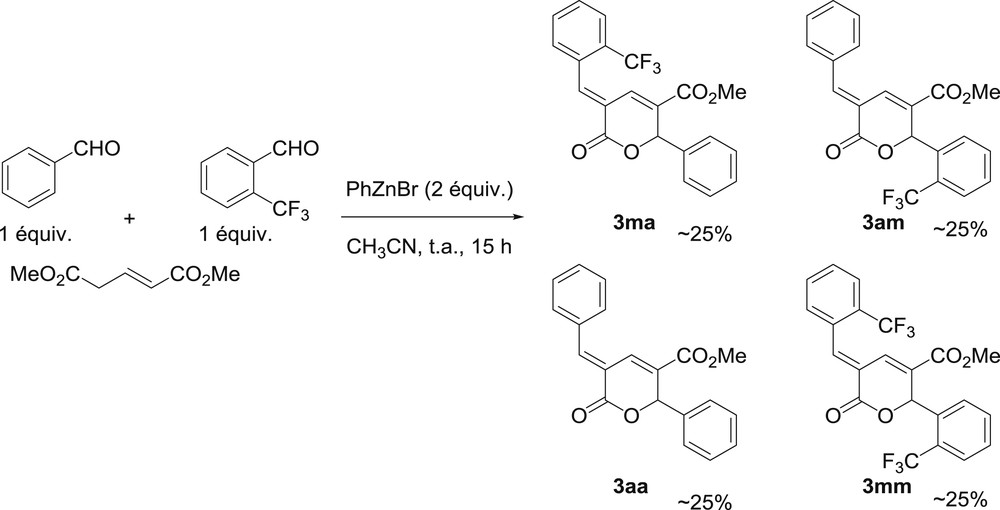
Essai de réaction utilisant deux aldéhydes différents.
3 Mécanisme réactionnel
Si l'on prend en compte le pKa de l'acétonitrile, qui est généralement rapporté autour de 25, et compte tenu de la génération possible d'arylzincique dans ce solvant sans protonation notable de cet organométallique, nous pouvons estimer le pKa du couple arylzincique/arène comme inférieur à 25. De plus, comme les arylzinciques sont facilement protonés par l'eau et les alcools (pKa inférieur à 20), nous pouvons affiner l'estimation du pKa du couple arylzincique/arène dans la gamme 20–25. Par conséquent, compte tenu de la capacité prévisible de l'arylzincique à déprotoner à la fois les alcools et les composés dicarbonylés, un mécanisme réactionnel possible peut être décrit comme suit (Schéma 4) :
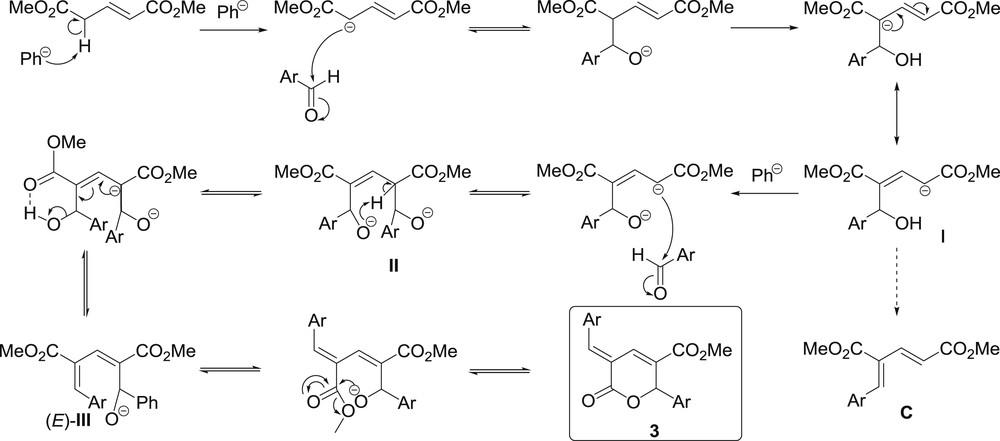
Mécanisme réactionnel supposé.
Le bromure de phénylzinc déprotonerait le glutaconate de diméthyle. Le carbanion résultant produirait alors une attaque nucléophile initiale sur l'aldéhyde suivie d'un échange intramoléculaire de proton conduisant à I. À ce stade, une deuxième déprotonation clé par l'espèce arylzincique se produirait. Cette déprotonation, probablement rapide, éviterait l’élimination intramoléculaire de l'hydroxyde conduisant à un composé conjugué C non réactif. Une seconde réaction d'aldolisation se produirait alors, avec formation d'un double alcoolate intermédiaire II qui perdrait l'hydroxyde par élimination intramoléculaire stéréosélective et conduirait à l'alcoolate (E)-III. La rotation vers le conformère s-trans et la cyclisation finale par transestérification intramoléculaire conduiraient à la lactone 3. On peut noter la remarquable stéréochimie E de la double liaison exocyclique [13], ce qui laisserait supposer que la réaction d’élimination d'hydroxyde s'effectue sous contrôle thermodynamique.
Le mécanisme décrit permet d'expliquer le comportement différent du para-diméthylaminobenzaldéhyde. En effet, on peut s'attendre à ce que la fonction diméthylamino, fortement donneuse, puisse agir à distance comme un groupe participant, ce qui favoriserait le départ rapide de l'hydroxyde au stade de I4 et entraînerait rapidement la formation du composé conjugué C4, mettant ainsi fin au processus réactionnel menant à la lactone 3 correspondante (Schéma 5).
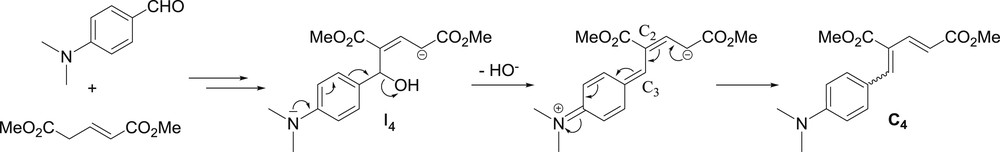
Participation d'un groupe diméthylamino distant pour la formation de 4.
On peut noter que, dans ce cas, la formation de la nouvelle double liaison n'est pas aussi stéréosélective que pour la génération de 3, car l'on obtient un rapport stéréoisomérique de ∼3/1. Ce résultat est plutôt logique. En effet, comme l'hydroxyde a déjà quitté l'intermédiaire II4 avant la formation de la double liaison, aucune participation de groupe voisin n'est plus envisageable. Ainsi, une rotation de la simple liaison C2C3 peut se produire, conduisant ainsi à des mélanges de stéréoisomères.
4 Conclusion
En conclusion, ce travail montre qu'il est possible de réaliser des pseudo-réactions à trois composants entre le glutaconate de diméthyle et des aldéhydes aromatiques afin de former des dihydropyranones 2,3,5-trisubstituées. La réaction est provoquée par la présence d'une base telle que le bromure de phénylzinc, qui possède une réactivité de nucléophile limitée vis-à-vis des aldéhydes aromatiques. Le mécanisme réactionnel proposé met en jeu un processus domino original de type aldolisation/aldolisation/élimination/lactonisation. La réaction semble générale, même si des aldéhydes substitués en position para par des groupements à effet mésomère donneur fort auraient tendance à fournir un produit conjugué intermédiaire au lieu de la lactone attendue.
5 Partie expérimentale
5.1 Données générales
Toutes les réactions ont été effectuées sous atmosphère d'argon. Les réactifs et solvants commerciaux ont été utilisés sans purification supplémentaire. Toutes les réactions ont été suivies par chromatographie en phase gazeuse (CPG) sur un chromatographe Shimadzu GC-2025 en utilisant une colonne Phenomenex ZB de 5 m. Les points de fusion (PF) ont été déterminés sur un appareil de fusion capillaire Büchi B-545 et ne sont pas corrigés. Les spectres infrarouge ont été enregistrés sur un spectromètre à transformée de Fourrier Brücker Tensor en mode ATR. Les spectres RMN ont été réalisés sur un spectromètre Brücker Avance II 400 dans CDCl3, à 400 MHz (1H), 100 MHz (13C) et 376 MHz (19F). Les déplacements chimiques (δ) sont donnés en parties par million (ppm) par rapport au signal du solvant non deutéré résiduel. Les multiplicités apparentes sont notées comme suit : s (singulet); d (doublet); t (triplet); q (quadruplet); m (multiplet); dd (doublet de doublets). Les chromatographies flash ont été réalisées sur silice (particules de 35 à 70 μm, 550 mesh). Les analyses de spectrométrie de masse haute résolution (SMHR) ont été réalisées par la plateforme analytique de l’ICOA (université d’Orléans) en mode d'ionisation electrospray (ESI). La structure cristalline du composé 3a a été déterminée au service de cristallochimie de l’Institut de chimie des substances naturelles, situé à Gif-sur-Yvette (UPR 2301).
5.2 Mode opératoire typique
Un ballon de 100 mL équipé d'un septum est purgé à l'argon puis de l'acétonitrile (40 mL), du zinc en poudre (6 g), de l'acide trifluoroacétique (0,2 mL) et du 1,2-dibromoéthane (0,4 mL) sont introduits sous vive agitation (∼500 tr/min). Le mélange est chauffé entre 50 °C et 70 °C jusqu’à dégagement d’éthylène, puis laissé à refroidir sous agitation continue jusqu’à la température de 20 à 30 °C (environ 10 min sont nécessaires). De la 2,2′-bipyridine (500 mg, 3,2 mmol), du bromure d'aryle (30 mmol) et du bromure de cobalt anhydre (700 mg, 3,2 mmol) sont ensuite ajoutés au mélange, qui est agité à température ambiante pendant 40 min supplémentaires. L'agitation est ensuite arrêtée et la solution surnageante prélevée à l'aide d'une seringue. La solution est ensuite placée dans un autre ballon de 100 mL sous atmosphère d'argon. Du glutaconate de diméthyle (3 mL, 21 mmol) et du benzaldéhyde (1,5 mL, 15 mmol) sont ajoutés successivement, et la solution est laissée sous agitation à température ambiante pendant 15 h. La solution réactionnelle est versée dans 150 mL d'une solution saturée de chlorure d'ammonium puis extraite par 2 × 100 mL d'acétate d’éthyle. Les fractions organiques sont regroupées et lavées par 100 mL d'une solution saturée de chlorure d'ammonium puis 100 mL d'une solution saturée de chlorure de sodium. La solution est séchée sur sulfate de sodium, puis le solvant est évaporé sous vide. L'huile brute est chromatographiée sur silice en éluant avec un mélange éther de pétrole/éther diéthylique de polarité croissante (90/10 à 50/50) pour fournir la lactone 3 attendue.
5.3 Données analytiques
5.3.1 (E)-5-benzylidène-6-oxo-2-phényl-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3a)
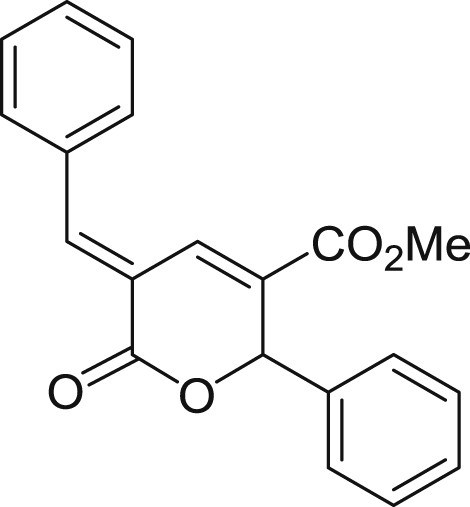
Préparé selon le mode opératoire général, à partir du benzaldéhyde (1,5 mL, 15 mmol). Le composé 3a est obtenu sous forme de poudre blanc cassé (1,81 g, 75%), PF = 147 °C.
RMN 1H (CDCl3) : δ = 8,17 (s, 1H), 8,01 (d, J = 0,8 Hz, 1H), 7,60–7,45 (m, 5H), 7,45–7,31 (m, 5H), 6,42 (s, 1H), 3,76 (s, 3H); RMN 13C (CDCl3) : δ = 164,3, 164,0, 145,2, 138,1, 134,0, 131,3, 130,7, 130,5, 129,2, 129,1 (2 signaux), 129,0, 127,2, 120,5, 79,8, 52,4; IR : ν = 2949,38, 1707,37, 1582,97, 1433,60, 1260,26, 1126,56, 1011,76, 837,37, 764,81, 690,90 cm−1; SMHR : m/z calculé pour C20H16NaO4 [M+Na]+ : 343,094080; trouvé : 343,094273.
5.3.2 (E)-5-(2-methylbenzylidène)-6-oxo-2-(o-tolyl)-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3b)
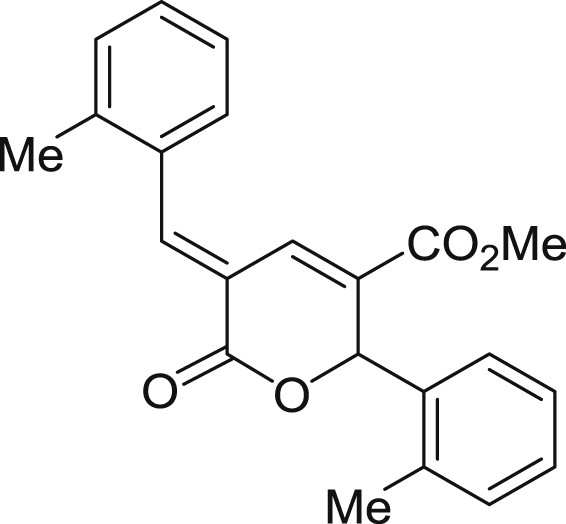
Préparé selon le mode opératoire général, à partir de l'ortho-tolualdéhyde (1,75 mL, 15 mmol). Le composé 3b est obtenu sous forme de poudre blanc cassé (1,15 g, 44%), PF = 152 °C.
RMN 1H (CDCl3) : δ = 8,30 (s, 1H), 7,98–7,81 (m, 1H), 7,49–7,31 (m, 4H), 7,30–7,26 (m, 2H), 7,24–6,99 (m, 2H), 6,70 (s, 1H), 3,71 (s, 3H), 2,64 (s, 3H), 2,43 (s, 3H); RMN 13C (CDCl3) : δ = 164,4, 163,7, 144,5, 138,5, 137,7, 135,7, 133,1, 132,0, 131,4, 130,9, 130,4, 129,7, 129,4, 128,6, 126,5, 126,2, 126,0, 121,1, 76,7, 52,3, 20,2, 19,2; IR : ν = 2950,41, 1707,21, 1432,03, 1267,34, 1211,77, 1124,93, 1013,97, 764,15 cm−1; SMHR : m/z calculé pour C22H20NaO4 [M+Na]+: 371,125380; trouvé : 371,125499.
5.3.3 (E)-5-(4-methylbenzylidène)-6-oxo-2-(p-tolyl)-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3c)

Préparé selon le mode opératoire général, à partir du para-tolualdéhyde (1,75 mL, 15 mmol). Le composé 3c est obtenu sous forme de poudre jaune pâle après évaporation poussée du glutaconate résiduel (0,92 g, 35%), PF = 136 °C.
RMN 1H (CDCl3) : δ = 8,13 (s, 1H), 8,03 (s, 1H), 7,47 (d, J = 8,0 Hz, 2H), 7,39–7,24 (m, 4H), 7,19 (d, J = 7,9 Hz, 2H), 6,38 (s, 1H), 3,77 (s, 3H), 2,45 (s, 3H), 2,35 (s, 3H); RMN 13C (CDCl3) : δ = 164,5, 164,2, 145,1, 141,4, 139,1, 135,3, 131,5, 131,3, 130,6, 129,9, 129,7, 128,7, 127,2, 119,8, 79,6, 52,3, 21,6, 21,2; IR : ν = 2954,22, 1702,64, 1589,81, 1258,77, 1120,15, 1004,84, 813,53 cm−1; SMHR : m/z calculé pour C22H20NaO4 [M+Na]+: 371,125380; trouvé : 371,125840.
5.3.4 (E)-5-(2-méthoxybenzylidène)-2-(2-méthoxyphényl)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3d)

Préparé selon le mode opératoire général, à partir de l'ortho-anisaldéhyde (2,05 g, 15 mmol). Le composé 3d est obtenu sous forme d'huile jaune collante (1,20 g, 42%).
RMN 1H (CDCl3) : δ = 8,36 (s, 1H), 7,88 (s, 1H), 7,52–7,18 (m, 4H), 7,17–6,81 (m, 4H), 6,58 (s, 1H), 3,89 (s, 3H), 3,78 (s, 3H), 3,69 (s, 3H); RMN 13C (CDCl3) : δ = 164,6, 164,5, 158,6, 157,9, 140,1, 132,1, 131,9, 130,8, 130,7, 129,6, 126,8, 125,2, 123,4, 121,1, 120,6, 120,5, 111,3, 111,2, 77,6, 55,6, 55,2, 52,1; IR : ν = 2950,81, 2839,60, 1709,98, 1640,62, 1596,43, 1248,11, 1021,46, 908,69, 725,85 cm−1; SMHR : m/z calculé pour C22H20NaO6 [M+Na]+: 403,115209; trouvé : 403,115081.
5.3.5 (E)-5-(3-méthoxybenzylidène)-2-(3-méthoxyphényl)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3e)
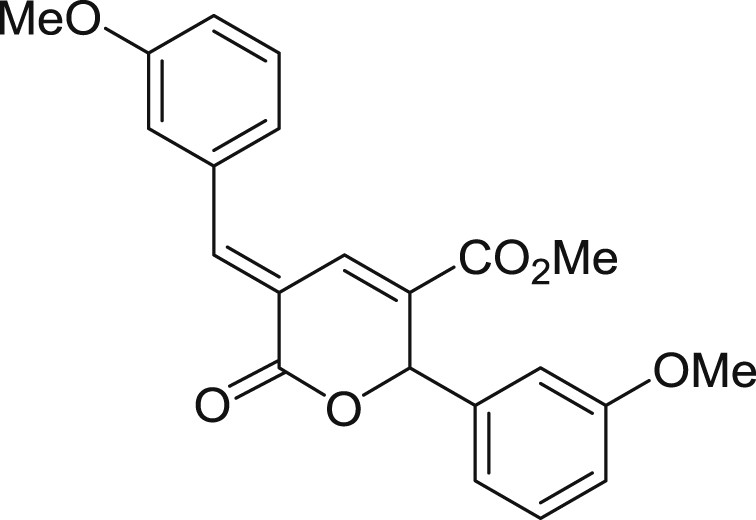
Préparé selon le mode opératoire général, à partir du méta-anisaldéhyde (1,8 mL, 15 mmol). Le composé 3e est obtenu sous forme de poudre blanc cassé (2,28 g, 80%), PF = 112 °C.
RMN 1H (CDCl3) : δ = 8.10 (s, 1H), 8.01 (d, J = 0.8 Hz, 1H), 7.46–7.38 (m, 1H), 7.33–7.25 (m, 1H), 7.12 (d, J = 7.7 Hz, 1H), 7.07–6.99 (m, 2H), 7.00–6.84 (m, 3H), 6,37 (s, 1H), 3,86 (s, 3H), 3,80 (s, 3H), 3,77 (s, 3H); RMN 13C (CDCl3) : δ = 164,3, 163,9, 159,9 (2 signaux), 145,0, 139,5, 135,2, 131,4, 130,1 (2 signaux), 129,0, 122,8, 120,7, 119,3, 116,4, 115,7, 114,5, 113,1, 79,6, 55,4, 55,3, 52,4; IR : ν = 3004,80, 2948,07, 2838,62, 1712,44, 1572,14, 1431,80, 1271,25, 1211,46, 1130,67, 1018,90, 874,39, 790,63 cm−1; SMHR : m/z calculé pour C22H20NaO6 [M+Na]+: 403,115209; trouvé : 403,115280.
5.3.6 (E)-5-(4-méthoxybenzylidène)-2-(4-méthoxyphényl)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3f)

Préparé selon le mode opératoire général, à partir du para-anisaldéhyde (1,8 mL, 15 mmol). Le composé 3f est obtenu sous forme d'huile jaune collante (1,14 g, 40%).
RMN 1H (CDCl3) : δ = 8,05 (s, 1H), 8,02 (s, 1H), 7,53 (d, J = 8,6 Hz, 2H), 7,30 (d, J = 8,7 Hz, 3H), 7,02 (d, J = 8,7 Hz, 2H), 6,88 (d, J = 8,7 Hz, 3H), 6,33 (s, 1H), 3,88 (s, 3H), 3,78 (s, 3H), 3,76 (s, 3H); RMN 13C (CDCl3) : δ = 164,5, 161,9, 160,2, 144,8, 132,7, 132,6, 131,6, 130,4, 128,7, 128,1, 126,7, 118,4, 114,7, 114,3, 79,3, 55,5, 55,3 (2 signaux) ; IR: ν = 2952,86, 2839,13, 1708,99, 1586,86, 1510,04, 1435,83, 1249,01, 1173,41, 1113,94, 1026,01, 830,52 cm−1; SMHR : m/z calculé pour C22H20NaO6 [M+Na]+: 403,115209; trouvé : 403,115187.
5.3.7 (E)-2-(furan-2-yl)-5-(furan-2-ylméthylène)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3g)
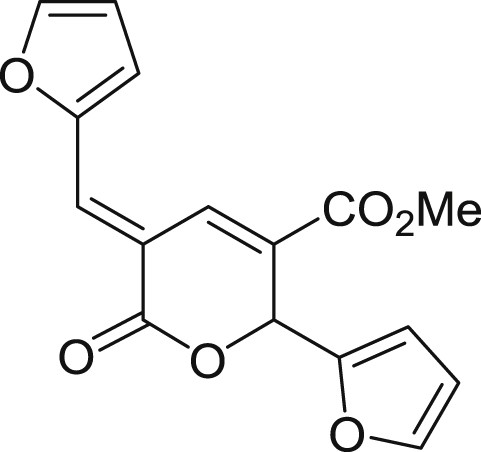
Préparé selon le mode opératoire général, à partir du 2-furaldéhyde (1,25 mL, 15 mmol). Le composé 3g est obtenu sous forme de poudre ocre (0,90 g, 40%), PF = 185 °C (décomposition).
RMN 1H (CDCl3) : δ = 8,56 (s, 1H), 7,77 (s, 1H), 7,66 (s, 1H), 7,39 (s, 1H), 6,93 (d, J = 3,3 Hz, 1H), 6,70–6,55 (m, 1H), 6,48–6,22 (m, 3H), 3,80 (s, 3H); RMN 13C (CDCl3) : δ = 164,3, 164,2, 151,5, 150,3, 147,8, 143,8, 133,5, 128,4, 125,0, 121,8, 115,8, 113,2, 110,7, 109,6, 72,3, 52,4; IR: ν = 2950,28, 1700,17, 1597,67, 1434,65, 1266,62, 1230,32, 1186,06, 1004,04, 752,63 cm−1; SMHR : m/z calculé pour C16H12NaO6 [M+Na]+: 323,052609; trouvé : 323,052651.
5.3.8 (E)-2-(thiophen-2-yl)-5-(thiophen-2-ylméthylène)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3h)

Préparé selon le mode opératoire général, à partir du 2-thiophènecarboxaldéhyde (1,4 mL, 15 mmol). Le composé 3h est obtenu sous forme de poudre jaune (0,90 g, 36%), PF = 138 °C (décomposition).
RMN 1H (CDCl3) : δ = 8,26 (d, J = 0,5 Hz, 1H), 8,16 (s, 1H), 7,74 (d, J = 5,0 Hz, 1H), 7,53 (d, J = 3,6 Hz, 1H), 7,38–7,17 (m, 2H), 7,10 (d, J = 3,5 Hz, 1H), 6,98 (dd, J = 4,9, 3,7 Hz, 1H), 6,61 (s, 1H), 3,86 (s, 3H); RMN 13C (CDCl3) : δ = 164,3, 164,1, 141,1, 137,4, 136,8, 136,2, 133,4, 131,3, 128,5, 128,4, 127,3, 127,1 (2 signaux), 116,7, 74,1, 52,6; IR : ν = 3110,35, 2947,33, 1703,86, 1576,48, 1264,58, 1183,21, 1114,35, 999,06, 709,65 cm−1; SMHR : m/z calculé pour C16H12NaO4S2 [M+Na]+: 355,006922; trouvé : 355,007072.
5.3.9 (E)-5-(3-fluorobenzylidène)-2-(3-fluorophényl)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3i)
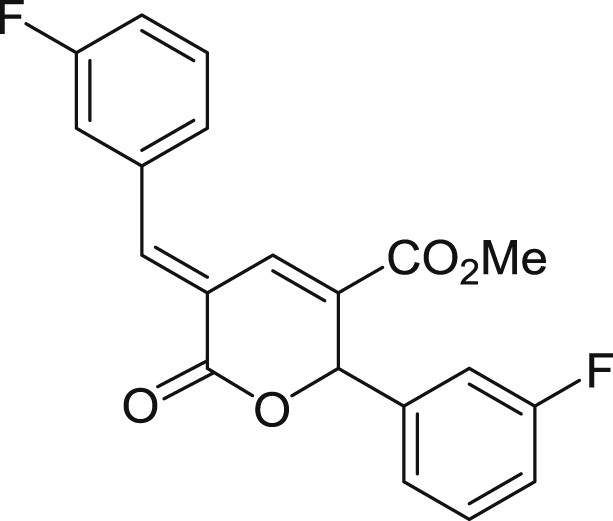
Préparé selon le mode opératoire général, à partir du 3-fluorobenzaldéhyde (1,6 mL, 15 mmol). Le composé 3i est obtenu sous forme de poudre blanc cassé (2,01 g, 75%), PF = 120 °C.
RMN 1H (CDCl3) : δ = 8.11 (s, 1H), 7.95 (d, J = 0.7 Hz, 1H), 7.54–7.45 (m, 1H), 7.41–7.27 (m, 2H), 7.26–7.14 (m, 3H), 7.11–7.02 (m, 2H), 6,40 (s, 1H), 3,79 (s, 3H); RMN 13C (CDCl3) : δ = 164,0, 163,2, 162,9 (d, J = 247,7 Hz), 162,8 (d, J = 248,2 Hz), 143,9 (d, J = 2,5 Hz), 140,3 (d, J = 6,8 Hz), 135,8 (d, J = 7,8 Hz), 130,9, 130,8 (d, J = 8,4 Hz), 130,7 (d, J = 8,3 Hz), 129,3, 126,2 (d, J = 3,0 Hz), 122,8 (d, J = 3,0 Hz), 121,1, 117,7 (d, J = 21,3 Hz), 116,9 (d, J = 22,2 Hz), 116,3 (d, J = 21,1 Hz), 114,2 (d, J = 22,5 Hz), 79,0 (d, J = 1,9 Hz), 52,6; RMN 19F (CDCl3) : δ = −111,28, −111,40; IR : ν = 3078,74, 2952,16, 1710,91, 1591,80, 1436,41, 1248,48, 1202,72, 1132,31, 1021,12, 795,77 cm−1; SMHR : m/z calculé pour C20H14F2NaO4 [M+Na]+: 379,075236; trouvé : 379,074746.
5.3.10 (E)-5-(4-fluorobenzylidène)-2-(4-fluorophényl)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3j)

Préparé selon le mode opératoire général, à partir du 4-fluorobenzaldéhyde (1,6 mL, 15 mmol). Le composé 3j est obtenu sous forme de poudre blanc cassé (2,03 g, 76%), PF = 115 °C.
RMN 1H (CDCl3) : δ = 8.10 (s, 1H), 7.95 (d, J = 0.8 Hz, 1H), 7.58–7.50 (m, 2H), 7.40–7.31 (m, 2H), 7.25–7.16 (m, 2H), 7.12–7.00 (m, 2H), 6,38 (s, 1H), 3,77 (s, 3H); RMN 13C (CDCl3) : δ = 164,2, 164,0 (d, J = 253,7 Hz), 163,6, 163,0 (d, J = 248,7 Hz), 144,1, 134,0 (d, J = 3,3 Hz), 132,6 (d, J = 8,7 Hz), 131,0, 130,1 (d, J = 3,4 Hz), 129,1 (d, J = 8,5 Hz), 129,0, 120,0 (d, J = 1,5 Hz), 116,5 (d, J = 22,0 Hz), 116,0 (d, J = 21,8 Hz), 79,0, 52,5; RMN 19F (CDCl3) : δ = −107,65, −111,93; IR: ν = 2954,56, 1714,87, 1597,70, 1505,58, 1260,27, 1224,46, 1134,86, 1023,61, 840,05 cm−1; SMHR : m/z calculé pour C20H14F2NaO4 [M+Na]+: 379,075236; trouvé : 379,075391.
5.3.11 (E)-5-(3-chlorobenzylidène)-2-(4-chlorophényl)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3k)
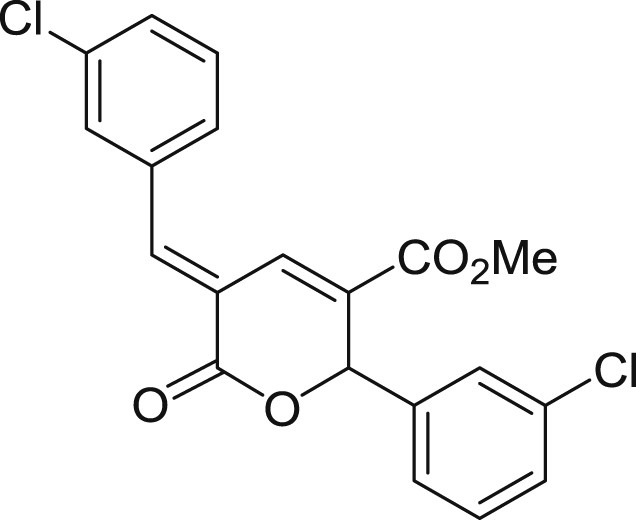
Préparé selon le mode opératoire général, à partir du 3-chlorobenzaldéhyde (1,7 mL, 15 mmol). Le composé 3k est obtenu sous forme d'huile incolore collante après évaporation poussée du glutaconate résiduel (1,61 g, 55%).
RMN 1H (CDCl3) : δ = 8,08 (s, 1H), 7,93 (s, 1H), 7,57–7,15 (m, 8H), 6,37 (s, 1H), 3,79 (s, 3H); RMN 13C (CDCl3) : δ = 164,0, 163,0, 143,8, 139,8, 135,5, 135,2, 134,9, 130,9, 130,6, 130,4 (2 signaux), 130,1, 129,5, 129,2, 128,3, 127,4, 125,3, 121,1, 79,1, 52,6; IR : ν = 2951,91, 1710,49, 1637,89, 1595,08, 1434,15, 1253,89, 1190,85, 1128,95, 683,04 cm−1; SMHR : m/z calculé pour C20H14Cl2NaO4 [M+Na]+: 411,016135; trouvé : 411,016137.
5.3.12 (E)-5-(4-bromobenzylidène)-2-(4-bromophényl)-6-oxo-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3l)
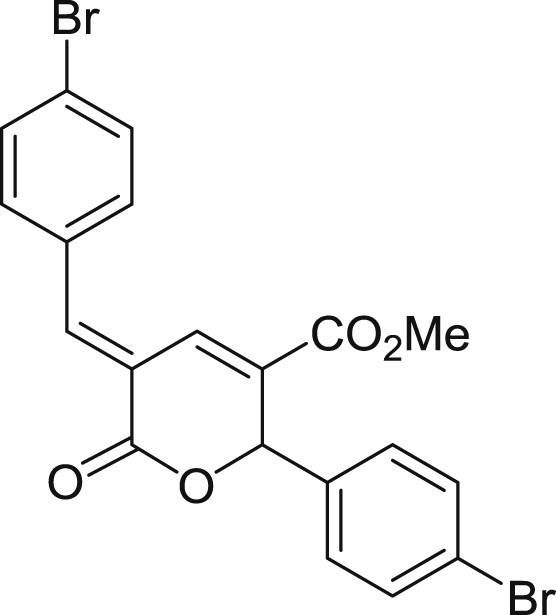
Préparé selon le mode opératoire général, à partir du 4-bromobenzaldéhyde (2,77 g, 15 mmol). Le composé 3l est obtenu sous forme de poudre blanc cassé (1,36 g, 38%), PF = 178 °C.
RMN 1H (CDCl3) : δ = 8,07 (s, 1H), 7,92 (s, 1H), 7,66 (d, J = 8,3 Hz, 2H), 7,52 (d, J = 8,4 Hz, 2H), 7,39 (d, J = 8,3 Hz, 2H), 7,25 (d, J = 8,3 Hz, 2H), 6,36 (s, 1H), 3,78 (s, 3H); RMN 13C (CDCl3) : δ = 164,1, 163,3, 144,1, 137,0, 132,7, 132,5, 132,2, 131,8, 130,9, 129,1, 128,8, 125,5, 123,5, 120,5, 79,1, 52,6; IR : ν = 2951,05, 1715,22, 1647,68, 1598,15, 1484,25, 1247,55, 1229,54, 1130,46, 1067,12, 1006,90, 838,43, 746,21 cm−1; SMHR : m/z calculé pour C20H15Br2O4 [M+H]+: 476,933160; trouvé : 476,933606.
5.3.13 (E)-6-oxo-5-(2-(trifluorométhyl)benzylidène)-2-(2-(trifluorométhyl)phényl)-5,6-dihydro-2H-pyran-3-carboxylate de méthyle (3m)

Préparé selon le mode opératoire général, à partir du 2-(trifluorométhyl)benzaldéhyde (2,0 mL, 15 mmol). Le composé 3m est obtenu sous forme de poudre blanche (2,26 g, 66%), PF = 169 °C.
RMN 1H (CDCl3) : δ = 8,39 (s, 1H), 7.89–7.69 (m, 4H), 7.67–7.43 (m, 4H), 7,35 (d, J = 7,7 Hz, 1H), 6,83 (s, 1H), 3,64 (s, 3H); RMN 13C (CDCl3) : δ = 163,6, 161,7, 142,4, 135,6, 132,7, 132,2, 132,1 (q, J = 1,9 Hz), 131,4, 130,7, 129,9, 129,7, 129,4 (q, J = 30,6 Hz), 129,2 (q, J = 31,0 Hz), 129,0, 127,7, 127,0 (q, J = 5,7 Hz), 126,7 (q, J = 5,2 Hz), 123,9 (q, J = 274,5 Hz), 123,7 (q, J = 273,9 Hz), 122,5, 75,7 (q, J = 2,4 Hz), 52,4; RMN 19F (CDCl3) : δ = −57,81, −60,29; IR : ν = 2959,25, 1713,23, 1310,05, 1267,73, 1106,66, 1016,78, 767,74 cm−1; SMHR : m/z calculé pour C22H14F6NaO4 [M+Na]+: 479,068849; trouvé : 479,068637.
Remerciements
Les auteurs remercient le CNRS et l’UPEC pour le financement de ces travaux.