

1 Introduction
Alkylaminophenols constitute a principle group of N,O-ligands [1]. They are vastly applied for coordination of first row transition complexes, serving, for example, as catalysts in oxidation [2–5], polymerization processes [6], or organic carbonate synthesis from CO2 [7–9]. Their benefits come from the tripod geometry of the alkylamine skeleton that allows for the formation of a chelating structure [10,11]. The amine nitrogen atom opens up the possibility of symmetry control [12]. Additionally, the electronic character of the nitrogen electron pair could be tuned and therefore fitted to many hard acids, such as high-valent d-block metals [13]. On the other hand, the presence of a phenol moiety in close vicinity of the nitrogen atoms secures the systems' high affinity to transition metals [14]. These features have attracted the attention of coordination chemists, who often impose additional factors on these ligands. One such structural factor is bulkiness of the substituents which allows for selective interactions (or even their elimination) with other molecules. Moreover, the recent enthusiasm in the idea of responsive molecules has launched the quest for systems which may be capable of covalent bond formation and cleavage within the molecule upon activation by an external stimulus [15]. This concept has already been investigated among bioresponsive contrast agents that belong to rapidly developed molecular probes [16,17]. Alkylaminophenols perfectly fit the basic elements of responsive molecules delivering a very tunable skeleton with both electronic and structural features, as has been demonstrated for magnetic resonance imaging contrast agents based on iron complexes [18–20]. Despite these benefits which are well appreciated due to the above-mentioned applications and despite some efficient methods such as the one published by Kerton et al that was carried out “on water” [21], synthetic approaches are still often a barrier to obtaining systems with the most desired features. For this reason, we present our study on branched, bulky N,N-bis(hydroxybenzyl)-N’-(pyridylmethyl)propanediamines 3 with one free nitrogen site for further substitution. This is an extended system of simpler N,N-bis(hydroxybenzyl)-N-ethan-N’-(pyridylmethyl)diamines which has already gained reasonable attention and has achieved most of the properties in groups of magnetic resonance imaging contrast agents [18].
2 Results and discussion
The approach to the synthesis of N,N-bis(hydroxybenzyl)-N’-(pyridylmethyl)propanediamines was initially launched by a method our group published for their N-ethyl counterparts [18] (Scheme 1). However, the major product separated for the post-reaction mixture was reduced salicylaldehyde. A plausible explanation for this reaction's failure was lower nucleophilicity of 3-aminopropan-1-ol due to internal H-bonding as compared to 2-aminoethanol.
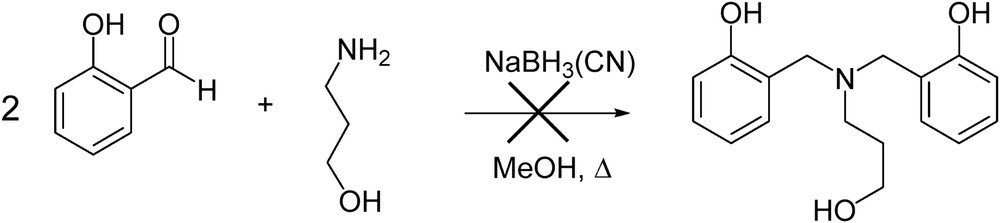
Synthesis of N,N-bis(hydroxybenzyl)-N-propanamines via reductive amination.
The alternative approach to the N,N-bis(hydroxybenzyl)-N-propane skeleton is via the Mannich reaction, analogous to ethyl derivatives [22]. There, due to the low regioselectivity of the benzene ring substitution in phenols, it is favored to block one of the ortho and the only para position with alkyl substituents. This was additionally beneficial to our idea of constructing a molecule with bulky substituents, thus, we used 2,4-dialkylphenols in the Mannich reaction (Scheme 2).

The Mannich reaction of 2,4-disubstituted phenols to N,N-bis(hydroxybenzyl)-N-propanamines. Yields of pure products are given in parentheses.
We were surprised to note that the yields (up to 66% for 1b), although generally not high, were much more elevated than in our previous reductive amination approach (34% [18]). The final yields refer to the pure product of an apparently five-molecular, multistep, two-stage Mannich reaction. Byproducts were easily separated from the post-reaction mixture and identified as structures with a single phenyl fragment (Scheme 3).
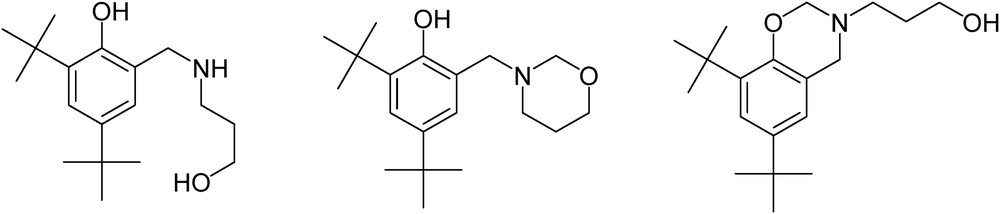
Byproducts formed in the Mannich reaction of 2,4-di(tert-butyl)phenol with 3-aminopropan-1-ol and paraformaldehyde.
The next step consisted chlorination of hydroxyl groups with thionyl chloride. The reaction proceeded through a multiphase reaction mixture of N- and O-sulfonation intermediates and their salts. However, the final workup by hydrolysis in aqueous bicarbonate led to crude product 2, which is usually of sufficient quality for further transformations. The stability of the 3-chloropropan-1-amine moiety was surprisingly good, even to obtain a fine crystal for X-ray analysis (Fig. 1). We did not observe dealkylation of the fragment during its synthesis or further transformation—this is not common since analogous compounds without hydroxyl substituents on the benzene ring are known to form intramolecular ammonium salts that undergo decomposition by dealkylation processes (Scheme 4) [23,24].
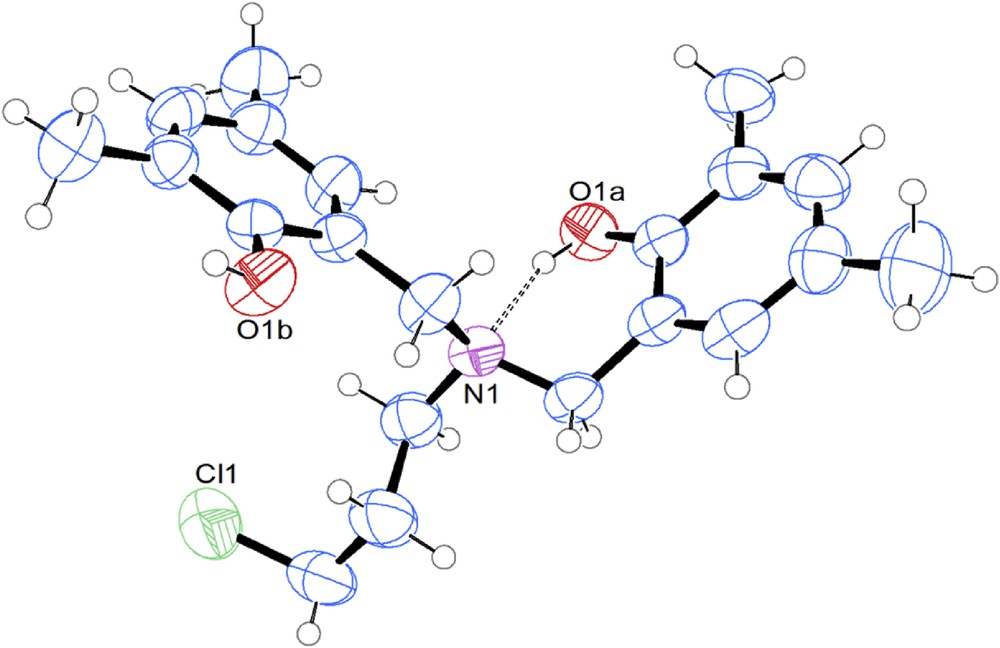
X-ray structure of 2a N,N-bis(2-hydroxy-3,5-dimethylbenzyl)-3-chloropropan-1-amine; 50% probability ellipsoids with hydrogen bonding presented by dashed lines.
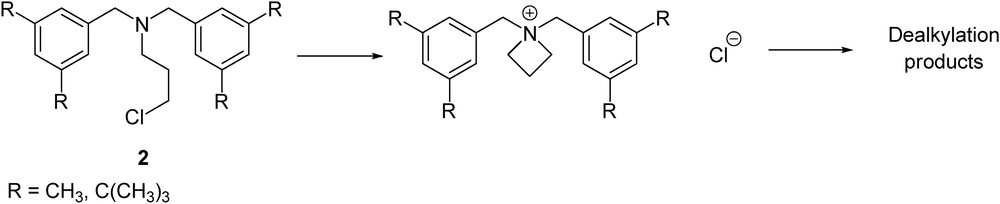
Dealkylation of non-hydrogen-bonded N,N-dibenzyl-N-chloroalkylamines.
The stability of our products could be explained by diminished nucleophilicity of the nitrogen atom due to the formation of hydrogen bonds. Such bonding is well identified in the X-ray structure (Fig. 1) with the N…HO distance equal to 1.884 Å, thus placing it in the classical H-bonding range. This interaction has been observed for related structurally characterized compounds [21,25,26].
The final step of merging the N,N-bis(hydroxybenzyl)-N-propanamine fragment with two additional nitrogen centers consisted in N-alkylation of 2-(aminomethyl)pyridine with chloroderivative 2. Our previous studies revealed that a large excess of 2-(aminomethyl)pyridine (at least 7-fold) was necessary to avoid its multiple alkylations. Moreover, adjusting the reaction conditions was important to increase the yield of the final product 3. It was found that the reaction time had to be adjusted to complete substrate 2 consumption (by thin-layer chromatography [TLC]). Prolongation of the reaction time led to serious product decomposition, thus diminishing its overall yield.
We also investigated the direct introduction of propane-1,3-diamine to the bis(hydroxybenzyl) skeleton. In this approach, we carried out the Mannich reaction between 2,4-di(tert-butyl)phenol, propane-1,3-diamine, and paraformaldehyde. While reductive elimination of salicylaldehyde and propane-1,3-diamine led to N,N′-bis(hydroxybenzyl)propane-1,3-diamine [19], the Mannich reaction led to cyclization and formation of N,N′-disubstituted 1,3-diazine 4, which was well identified by X-ray analysis (Scheme 5) [27,28]. Therefore, the route as described above from 3-aminopropan-1-ol via chlorination is a reasonable alternative that successfully leads to the desired N,N-bis(hydroxybenzyl)-N-propanamines.
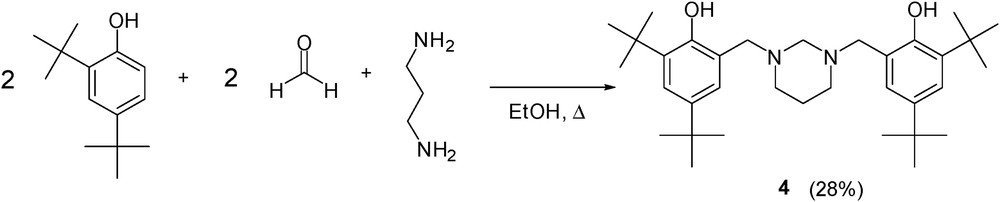
The Mannich reaction of 2,4-di(tert-butyl)phenol, propane-1,3-diamine, and paraformaldehyde.
We show a convenient and selective way of synthesis of bulky alkylaminophenols as a promising skeleton with a free site on the nitrogen atom for responsive systems and coordination compounds.
Acknowledgments
The Silesian University of Technology is acknowledged for the financial support.