

1 Introduction
Azasteroidal compounds target a variety of biological processes and therefore are potential candidates for the treatment of a large group of diseases, including breast and prostate cancers (PCas), benign prostatic hyperplasia (BPH), osteoporosis, and autoimmune diseases [1–4]. PCa is one of the most frequent malignant tumors in the world and is the second leading cause of death in men [5,6]. The androgen receptor (AR) signaling pathway plays an important role in PCa development and progression. In fact, the initial growth of prostate carcinomas is dependent on the androgens, testosterone and 5α-dihydrotestosterone (DHT), which is a metabolite of testosterone and the most potent natural androgen [7–9]. In the human body, testosterone is converted, in an irreversible way, into DHT by means of the 5α-reductase (5AR) enzymes [10]. Once DHT is formed, it binds to the AR, promoting its activation, which triggers the transcription of several genes. Therefore, because these steroids have a significant role in cell growth and survival, the inhibition of the androgenic pathway has been widely explored for the prevention and treatment of PCa and BPH [11]. In this context, several nitrogenated steroidal derivatives have been developed and are clinically used for the treatment of these conditions, including abiraterone acetate and finasteride (Fig. 1). These drugs reduce androgen levels through inhibition of 17α-hydroxylase/17,20-lyase (CYP17A1), an enzyme required for androgen biosynthesis, and inhibition of 5AR, respectively [12,13]. However, several drug-related adverse effects were described, including interferences in sexual function, which can limit their use. In addition, because of the risk of interactions with other drugs and to several contraindications, there is still a need to develop more potent and selective drugs for this purpose [14].
Examples of androgen biosynthesis inhibitors. Finasteride is a competitive inhibitor of 5α-reductase, whereas abiraterone is an inhibitor of CYP17A1.
Within the natural, semisynthetic, and synthetic steroidal derivatives with high antitumor interest [5,15], several other examples related to the present research work can be highlighted. In this context, some 17(E)-picolinylidene androstane derivatives were described as potential inhibitors of prostate and breast cancer cell growth. Interestingly, their antiproliferative activity against PC-3 cells is correlated with the antiproliferative effect observed with abiraterone and the predicted CYP17A1 binding affinities [4]. In other research studies, several 4-aza-d-homosteroids having purine nucleoside analogues bound to C17 were prepared and evaluated as antitumor agents in breast (MCF-7) and prostate (PC-3) cell lines. Interestingly, several of these compounds revealed to have potent antiproliferative effects against PC-3 cells [16–18]. Furthermore, the introduction of a 16-arylidene group into the steroid skeleton is also associated with important cytotoxic effects in several cell lines, being considered as a relevant pharmacophore for anticancer activity [19–24].
Bearing in mind all these facts, the general aim of the present work was the preparation of new 4-azasteroidal derivatives modified at C16 with aromatic and heteroaromatic rings and the evaluation of their effects on different cell lines. The structure of the synthesized compounds was validated through nuclear magnetic resonance (NMR), infrared (IR), and high-resolution mass spectroscopy (HRMS). The assessment of the antiproliferative effects of these compounds was performed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in LNCaP, PC-3, T47-D, and Normal Human Dermal Fibroblast (NHDF) cell lines as well as by flow cytometry after propidium iodide (PI) staining. In addition, their mode of interaction with estrogen receptor α (ERα), AR, CYP17A1, and 5β-reductase (5BR) was also studied by in silico molecular docking [25,26].
2 Results and discussion
2.1 Chemistry
The synthesis of 4-azaandrost-5-ene-3,17-dione has been carried out as depicted in Scheme 1. For this, testosterone was treated with pyridinium chlorochromate (PCC) to form androstenedione (1) in an excellent yield of 97%. Then, 1 was treated with sodium periodate and potassium permanganate to produce 2 in 83% yield by an oxidative cleavage reaction [27,28]. Finally, 4-azaandrost-5-ene-3,17-dione (3) was obtained from the azacyclization reaction of 2. Different procedures for this reaction involving different catalysts or reagents and energy sources (e.g., microwaves) are described [29,30]. Within these methods, the use of acetic acid and ammonium acetate proved to be a practical approach and allowed the preparation of the desired products in high yield, nevertheless the removal of acetic acid can be a difficult step. To overcome this problem, an extraction with a large portion of dichloromethane (DCM) followed by washing the organic phase several times to ensure a total remotion of the acetic acid present in the reactional mixture is required. IR and NMR spectroscopy data of product 3 were similar to that described in the literature [30–32].
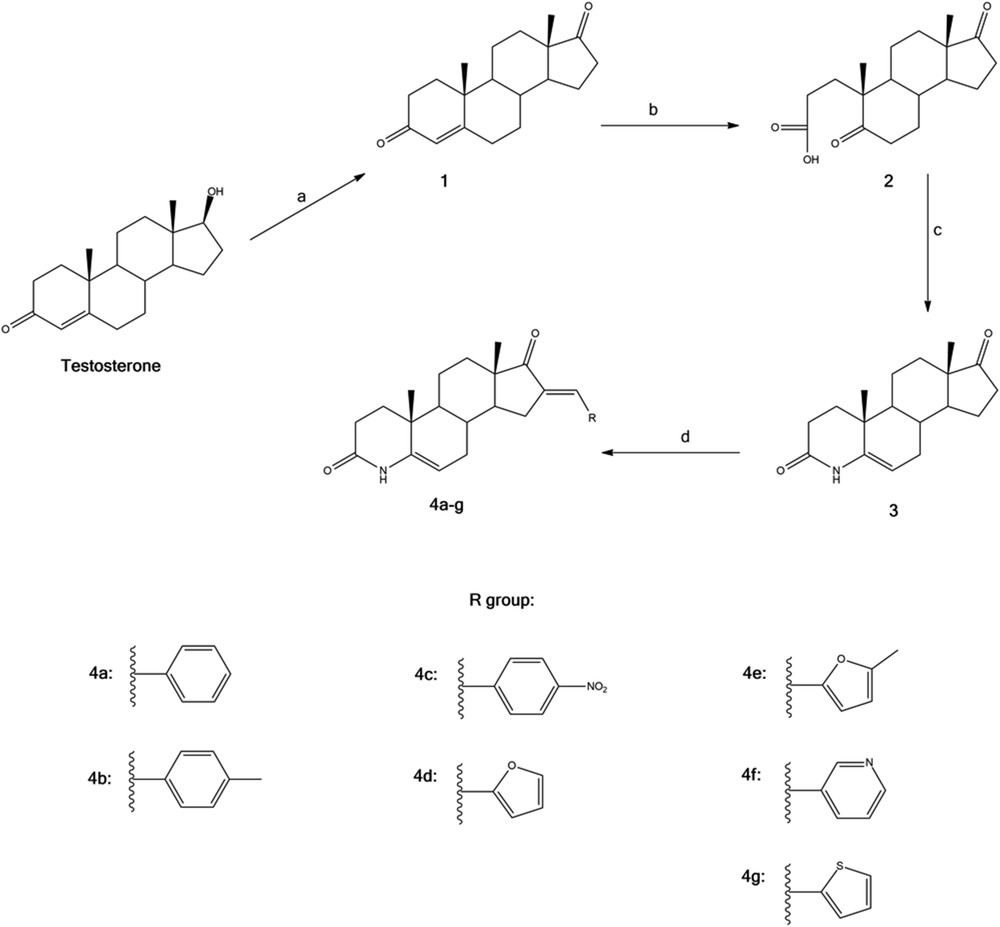
Synthesis of the compounds 4a–g from testosterone. Reagents, reaction conditions and yields: (a) PCC, DCM, rt, 2 h, 97% (b) NaIO4, KMnO4, Na2CO3, i-PrOH, reflux 3 h, 83%; (c) CH3COONH4, CH3COOH, reflux 4 h, 98%; (d) KOH, aldehyde, EtOH, rt, 12–24 h, 56–85%.
Modifications on the D-ring of steroids are important because such alterations frequently result in more effective receptor binding or increase in the bioavailability [31]. Considering the number of reported biological activities associated with structural analogues, the synthesis of novel 4-azasteroids with modifications on the D-ring by introducing an arylidene group was herein decided. In fact, the aldol condensation of 4-azaandrost-5-ene-3,17-dione with various aldehydes at room temperature in alkaline medium [20,22] afforded the corresponding arylidene derivatives 4a–g in very acceptable global yields (Table 1). To the best of our knowledge all these 16-arylidene-4-azaandrostenes are new compounds. The methine-bridge proton at 16-C of all arylidene derivatives 4a–g appeared at near 7.3 ppm in the 1H NMR spectra. The configuration of this new alkene with respect to the carbonyl at 17-C has been assigned E on the basis of earlier reports [21]. All spectral data of each compound were found to be consistent with the formation of the desired products.
New 16-arylidene-4-azaandrostene steroidal derivatives synthesized and global yields (%). (Hetero)arylidene in 4 was introduced through the aldol condensation at the C16 of 3 with respective aldehyde.
| Aldehyde | Product | Global yield (%) |
| Benzaldehyde | 4a | 44 |
| p-Tolualdehyde | 4b | 62 |
| 4-Nitrobenzaldehyde | 4c | 59 |
| Furaldehyde | 4d | 52 |
| 5-Methyl-2-furaldehyde | 4e | 67 |
| 3-Pyridinecarboxaldehyde | 4f | 62 |
| 2-Thiophenecarboxaldehyde | 4g | 66 |
2.2 Biology
2.2.1 Screening of cell proliferation effects
The action of the novel 4-azaandrostane derivatives 4a–g and their synthetic precursors (1, 2, and 3) was examined by the MTT assay on the proliferation of LNCaP, PC-3, T47-D, and NHDF cells as models of androgen-dependent, androgen-independent, and hormone-responsive breast cancers as well as nontumoral cells. Because of the structural similarities between these derivatives and finasteride, this drug was also included in the study. Moreover, 5-fluorouracil (5-FU) was also used in the assay as a clinically used antitumor positive control. The androgens, testosterone and DHT, were included in the assay as endogenous ligands of ARs to compare their effect on cell proliferation with the novel compounds. In a first assay, cells were exposed to all 4-azaandrostene derivatives at the concentration of 30 μM for 72 h, similarly as was performed in other studies [33–35]. The results of this screening are shown in Fig. 2. Compound 4a inhibited the proliferation of all tumoral cells, but not of normal fibroblasts. Compound 4c was toxic to variable extents in all cell lines. Interestingly, compound 4b originated a relevant reduction of cell proliferation in the androgen-dependent LNCaP cells to values lower than 50% of the negative control, presenting no toxicity in the fibroblast cell lines and reduced cytotoxicity in PC-3 cells. In T47-D cell line, compounds 4a and 4g also showed relevant antiproliferative effects. Finally, in NHDF cells, only compound 4c markedly reduced the cell proliferation. On the other hand, in this cell line, 4f and 4g seemed to stimulate the cell proliferation.
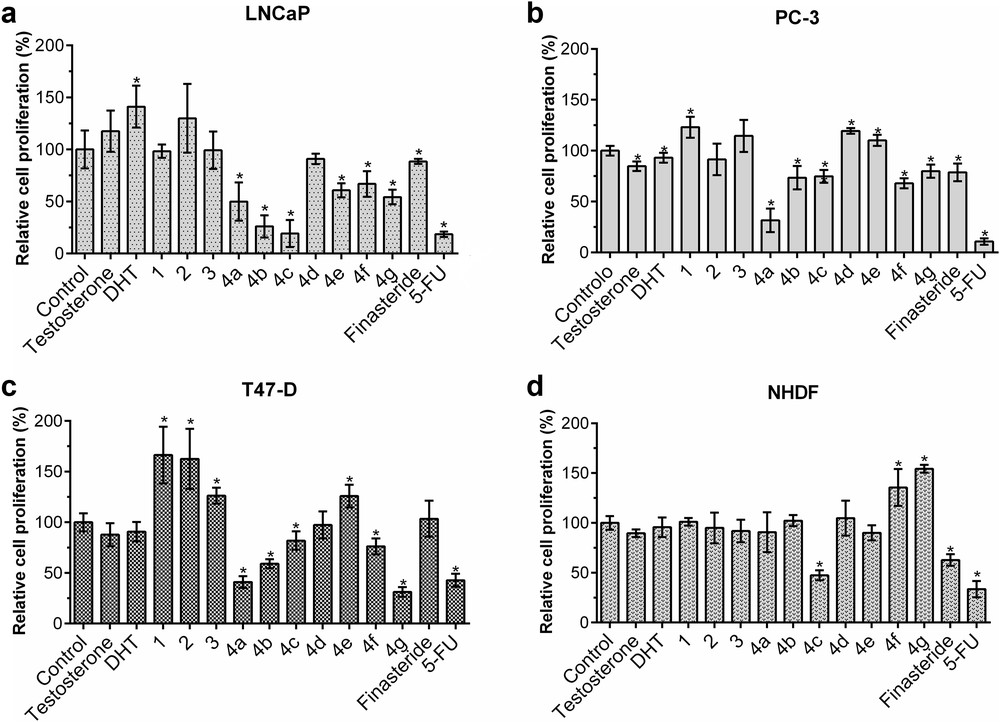
MTT screening results. Relative cell proliferation of (a) LNCaP, (b) PC-3, (c) T47-D, and (d) NHDF cells incubated with the compounds synthesized, substrates and finasteride, for 72 h of exposition at a concentration of 30 μM, determined by the MTT assay, spectrophotometrically quantifying formazan at 570 nm. Data are expressed as a percentage of cell proliferation in comparison with the negative control and are indicated as means ± SD and are representative of at least two independent experiments. ∗P < 0.05 vs the control (the Student t test).
In contrast, the intermediate products 1, 2, and 3 showed no significant effects on NHDF cells, as well as testosterone and DHT, although the percentage of LNCaP cell proliferation increased when exposed to DHT, as expected, probably because of its binding to AR [36]. In LNCaP cells, 1, 2, and 3 also did not show any significant effect but it seems to stimulate the proliferation of T47-D cells. In this cell line, testosterone and DHT showed no significant effect in cell proliferation.
After this preliminary evaluation, concentration–response studies were performed for the most antiproliferative compounds, finasteride and 5-FU in all cell lines, and the half maximal inhibitory concentration (IC50) of the tested compounds was estimated. The results obtained are shown in Table 2, and some relevant points can be highlighted. Interestingly, the data obtained confirmed the observed selectivity of these compounds in the screening results. In fact, the steroid 4b was selective for the hormone-dependent LNCaP cells (IC50 = 28.28 μM) when comparing with the PC-3 cell line (IC50 = 45.31 μM). Although 4c presented a higher IC50 value for LNCaP cells (IC50 = 37.20 μM), a clear selectivity for these cells was also observed. On the other hand and as expected, the potency of the steroid 4a is relatively similar in both PC-3 and LNCaP cells. Compound 4g, which showed relevant cytotoxicity only in T47-D cells, has an IC50 value of 16.34 μM, being the best value in this cell line. From these data, compound 4b appeared to be an interesting 16-arylidene-4-azaandrostene for more advanced studies, namely, in LNCaP cells.
Estimated IC50 values for various compounds in NHDF, LNCaP, PC-3, and T47-D cells.a
| Compound | R | NHDF | LNCaP | PC-3 | T47-D | ||||
| IC50 (μM) | r2 | IC50 (μM) | r2 | IC50 (μM) | r2 | IC50 (μM) | r2 | ||
| 4a | Image 1 | – | – | 24.30 | 0.82 | 22.63 | 0.98 | >100 | – |
| 4b | Image 2 | – | – | 28.28 | 0.82 | 45.31 | 0.89 | – | – |
| 4c | Image 3 | >100 | – | 37.20 | 0.84 | >100 | – | – | – |
| 4g | Image 4 | – | – | – | – | – | – | 16.34 | 0.96 |
| 5-FU | – | 9.16 | 0.87 | 1.71 | 0.97 | 2.30 | 0.96 | 2.89 | 0.98 |
| Finasteride | – | >100 | – | 70.95 | 0.95 | >100 | – | >100 | – |
a The cells were treated at various concentrations (0.01, 0.1, 1, 10, 50, and 100 μM) for 72 h. The antiproliferative effect was determined by the MTT assay, and the IC50 values were calculated by sigmoidal fitting. The data shown are representative of at least two independent experiments. The coefficient of determination (r2) is indicative of how closely the sigmoidal curve generated can be fitted with nonlinear regression statistics for the cell lines and incubation periods tested.
2.2.2 Characterization of the cytotoxic effect of compound 4b on LNCaP cells
After 24 and 72 h of exposition to the tested compounds, a microscopic cell morphology study was performed, and it was observed that cells treated with either 4b or finasteride showed mainly reduced cell density, with most cells keeping the healthy morphology (Supporting data). Cell viability of LNCaP cells when exposed to compounds 4b, finasteride, and 5-FU was then more accurately evaluated by flow cytometry quantification of PI permanent (dead) versus nonpermanent cells (live cells) (Fig. 3) [37]. Interestingly, arylidene steroid 4b induced a small reduction in the number of living cells similar to that observed with finasteride. This resemblance suggests that these compounds may have a similar mechanism of action in terms of cell death, expected to occur secondarily to the inhibition of cell cycle progression [38]. In parallel, it can be noted that after 72 h of incubation a relevant percentage of cells in R3, which is a region of less clear significance, was observed. This can be indicative of partial PI permeability or increased autofluorescence (early apoptotic cells) and/or cell debris with degraded DNA (advanced cell death stage) [37].
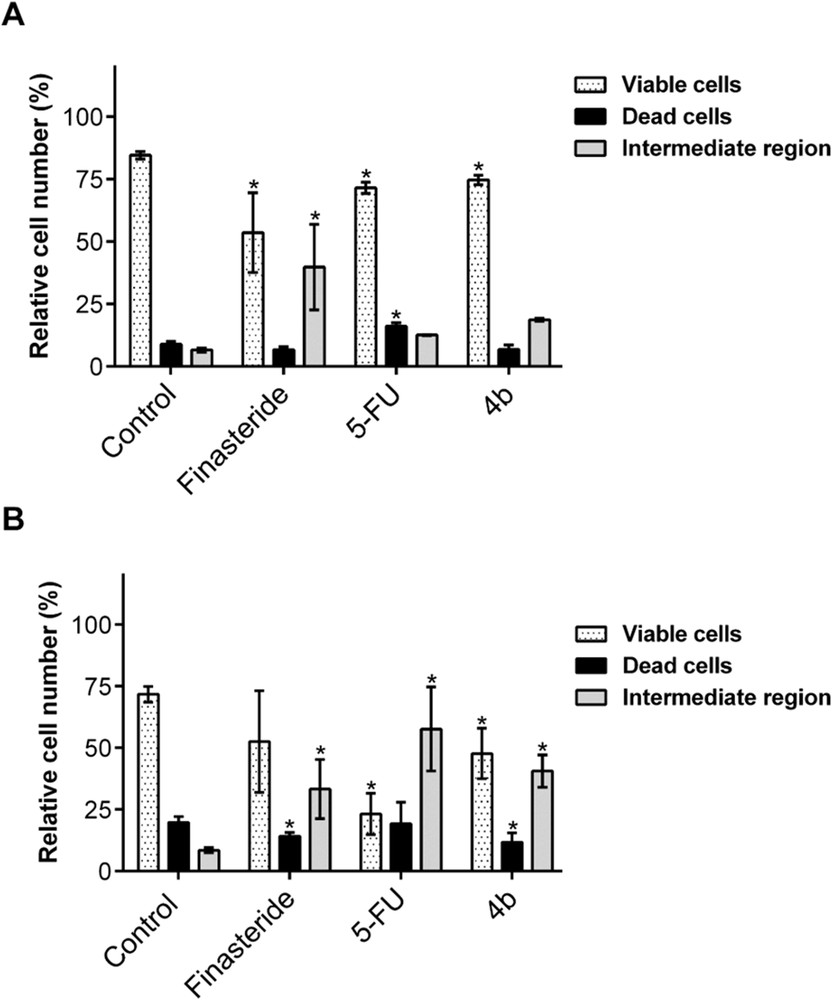
Percentage distribution of the events in the different regions R1, R2, and R3. (a) After 24 h and (b) 72 h of incubation. Data are expressed as a percentage of cells in the different regions indicated as means ± SD of 4–6 samples and are representative of at least two independent experiments. ∗P < 0.05 vs the control (one-way analysis of variance).
2.3 Molecular docking studies
To verify the potential affinity of these novel compounds to several proteins an in silico study using molecular docking was performed. This study aimed to evaluate the existence of potential interactions between these novel 4-azaandrostene derivatives and proteins, which are known to interact with steroids. For this, molecular docking simulations were performed against known targets of steroidal drugs currently used in the treatment of breast cancer, BPH, and PCa. Docking simulations were performed with 5BR, because the X-ray crystal structure of 5AR enzyme is not accessible yet [39]. Therefore, the crystal structure of 5BR was chosen, which is available and has a high homology level with 5AR. In this context, regardless of the potential selectivity problem, several studies used the 5BR crystal structure, as surrogate of 5AR, to perform the docking simulations [25,26]. The other protein targets were chosen for this study mainly according to the following criteria: (1) a high-resolution X-ray crystal structure is available in complex with a steroidal drug or other ligand and (2) the protein is a target of clinically approved steroid-based anticancer drugs in the treatment of hormone-dependent breast cancer or PCa. Thus, this study was performed for each arylidene azaandrostene derivative, 4a–g, not only against 5BR, but also against ERα, AR, human CYP17A1, and aromatase. This last enzyme was also included because it was identified in T47D cells [40] and because several 16E-arylidene androstene derivatives have aromatase inhibitory activity [22]. Three-dimensional structural coordinates of protein receptors were obtained from the protein data bank (PDB) and molecular docking was performed using the program AutoDockTools. To validate the docking method, simulations were carried out between crystallized ligands/drugs with the respective proteins and all control redocking simulations were able to reproduce the ligand–protein interaction geometries presented in the respective crystal structures with a root-mean-square distance (RMSD) ≤2.0 Å. The results of redocking are shown in Table 3 and Fig. S5 (Supporting data), and it can be observed that all simulations exhibit an RMSD <1.0 Å. On the basis of the control redocking simulations, predicted binding energies are analyzed in comparison with the value observed for the control. From this docking study, as it can be seen in Table 3, some relevant binding energies were predicted (see also Tables S1–S5, Supporting data). In fact, the results relatively to 5BR revealed that compounds 4b, 4c, 4e, and 4f have lower energies than the control, finasteride. These results showed that there is a possibility of these synthesized compounds to be potential 5AR inhibitors. On the other hand, the calculated affinity of these 4-azasteroid derivatives synthesized from testosterone to ERα was lower than that observed with 17β-estradiol. Moreover, when the molecular docking was performed with AR it was observed that compound 4d showed higher affinity than DHT. The simulations with aromatase revealed that there is, generally, a low affinity of these new compounds to this enzyme. Given the typical selectivity problem of this type of compounds, this result is interesting. Finally, considering the docking results against CYP17A1, when compared with abiraterone, the affinity of the tested compounds is lower. However, it is interesting to notice the generally low values, which can be suggestive that these compounds can also have important interactions with this relevant target.
Predicted binding energies of 4-azaandrostene derivatives 4a–g calculated from molecular docking against known protein targets of steroidal molecules: 5BR, ERα, AR, CYP17A1, and aromatase.
| Compound | Autodock binding energy (kcal mol−1) | ||||
| 5BR | ERα | AR | CYP17A1 | Aromatase | |
| 4a | −9.47 | −8.87 | −9.62 | −11.31 | 0.70 |
| 4b | −9.99 | −7.49 | −6.76 | −11.37 | 3.58 |
| 4c | −10.08 | −7.90 | −2.61 | −11.13 | 5.45 |
| 4d | −9.81 | −9.15 | −11.78 | −11.01 | −6.43 |
| 4e | −10.00 | −8.81 | −10.36 | −10.96 | −4.83 |
| 4f | −9.97 | −8.80 | −10.16 | −11.18 | −6.72 |
| 4g | −9.87 | −9.09 | −10.77 | −11.46 | −6.73 |
| Finasteride | −9.90 | – | – | – | – |
| 17β-Estradiol | – | −9.28 | – | – | – |
| DHT | – | – | −11.20 | – | – |
| Abiraterone | – | – | – | −11.78 | |
| Androstenedione | – | – | – | – | −12.65 |
The main interactions between these macromolecules and the best scored compounds in molecular docking simulations are shown in Figs. S6, S8, and S10 (Supporting data). In addition, the overlap of 4d with DHT on AR binding pocket is presented in Fig. S7. The overlap of compound 4b with finasteride on the active site of 5BR is also shown in Fig. S9. Through the analysis of these data, it is possible to conclude that the lactam group seems to be essential in establishing interactions with different amino acids of the studied proteins. In Fig. S9, it is possible to observe that lactam groups of the two compounds, 4b and finasteride, are aligned and the two compounds have the same general orientation. In addition, it is important to mention that as all compounds have the lactam substituent in common, the different binding energies in each protein are associated with the different arylidene groups. In this context, the effect of the presence of heteroaromatic rings on the binding energies to AR is evident.
In cell proliferation assays, it was possible to observe more relevant effects in androgen-dependent cells (LNCaP) than in the androgen-independent cells (PC-3). Considering the concomitant analysis of these docking simulations and cell proliferation assays as well as the structural similarity of these compounds with finasteride, it seems that the cell effects of these compounds can be related to their potential interaction with 5AR. However, their interference in other relevant points of the androgen biosynthesis and signaling pathways cannot be excluded, particularly their binding to CYP17A1, as referred. In T47-D cells, there is no evidence of a relationship between the cell proliferation effects of these compounds and their predicted affinities to ERα and aromatase enzyme.
3 Conclusions
In the present work, a successful synthesis of novel 4-azaandrost-5-ene-3,17-diones with a modified D-ring through the introduction of an aromatic or heteroaromatic aldehyde at C16 by a condensation reaction is described. Interesting results are observed in the MTT cell proliferation assay in LNCaP cells. In fact, the 4-azaandrostene derivatives 4a, 4b, and 4c presented relevant antiproliferative effects in this cell line and, within these, 4b presented the lowest calculated IC50 value (28.28 μM). In the androgen-independent PC-3 cell line, only 4a showed a relevant antiproliferative effect (IC50 = 22.63 μM), which suggests that most of these compounds can be interfering in the androgen pathway. The most cytotoxic compound on the breast cancer T47-D cells was the steroid 4g (IC50 = 16.34 μM) whereas finasteride had no relevant effect in this cell line. In addition, generally all 4-azaandrostane derivatives 4 seem to have low toxic effects on the normal cells (NHDF). Therefore, and considering these results, assessing the viability of LNCaP cells when exposed to compound 4b through a flow cytometry assay with PI staining was considered important. From these assays, a similar effect between finasteride and compound 4b can be observed. Molecular docking simulations showed that these novel 4-azaandrostene arylidene derivatives 4 can potentially inhibit 5BR, a surrogate of 5AR. In addition, with the exception of aromatase, they can also interact with other common targets of steroidal drugs, especially with CYP17A1. In conclusion, 4b seems to be the most promising selective antiproliferative agent against hormone-dependent prostatic cancer cells. Interestingly, a positive correlation between this cell effect and the 5AR inhibitory potential was observed.
4 Experimental section
4.1 Chemistry
4.1.1 General considerations
Reagents and solvents were purchased from standard sources and were purified and/or dried whenever necessary using standard procedures before use [41]. Finasterida Tetrafarma 5 mg was purchased from Tetrafarma – Produtos Farmacêuticos, Lda, Portugal, and finasteride was extracted from the tablets [42]. The reactions were performed under heating and magnetic stirring using Heidolph plates. Thin layer chromatography (TLC) analysis was performed using 0.20 mm Al-backed silica-gel plates (Macherey–Nagel 60 F254, Duren, Germany), and after elution the plates were visualized under UV radiation (254 nm) in a CN-15.LC UV chamber. Then a revelation step with an ethanol/sulfuric acid (95:5) mixture was performed, followed by heating at 120 °C. For the isolation and purification of product 1, a column chromatography was performed using silica gel (0.063–0.200 or 0.040–0.063 mm), acquired from Merck (NJ). The eluents used are indicated as a v/v proportion in the experimental procedure. The evaporation of solvents was achieved by using a rotary vacuum drier from Büchi (R-215). Attenuated total reflectance IR spectra were collected using a Thermo Scientific Nicolet iS10, smart iTR, equipped with a diamond attenuated total reflectance crystal. For IR data acquisition, each solid sample was placed onto the crystal and the spectrum was recorded. An air spectrum was used as a reference in absorbance calculations. The sample spectra were collected at room temperature in the 4000–600 cm−1 range by averaging 16 scans at spectral resolution of 2 cm−1.1H NMR and 13C NMR spectra were recorded using a Bruker Avance 400 MHz spectrometer (1H NMR at 400.13 MHz and 13C NMR at 100.62 MHz) and were processed with the software TOPSPIN (v. 3.1) (Bruker, Fitchburg, WI). Deuterated chloroform (CDCl3) was used as a solvent. Chemical shifts are reported in parts per million (δ) relative to TMS or solvent as an internal standard. Coupling constants (J values) are reported in hertz (Hz) and splitting multiplicities are described as s = singlet, d = doublet, t = triplet, combinations of above, or m = multiplet. ESI-TOF mass spectrometry was performed by the microanalysis service using a QSTAR XL instrument.
4.1.2 Preparation of precursors 1, 2, and 3
4.1.2.1 Androstenedione (1)
Testosterone (576.8 mg, 2 mmol) in DCM (90 mL) was treated with PCC (0.7186 g, 3.33 mmol). The resulting suspension turned from orange to brown and was stirred at room temperature for 2 h. The reaction mixture was diluted with 25 mL of diethyl ether and filtered through a pad of Celite. After removal of solvent, the brown oil was purified by column chromatography (ethyl acetate/petroleum ether 40–60 °C, 3:1) to give product 1 as a white solid (547.6 mg, 97%); mp 166–168 °C; IR (cm−1): 2918, 1731, 1659.1H NMR (CDCl3, 400 MHz) δ: 5.68 (1H, s, 4-H), 1.15 (3H, s, 19-CH3), 0.85 (3H, s, 18-CH3). 13C NMR (CDCl3, 101 MHz) δ: 220.35, 199.29, 170.28, 124.15, 53.83, 50.85, 47.51, 38.65, 35.75, 35.16, 33.91, 32.57, 31.29, 30.76, 21.75, 20.32, 17.39, 13.71.
4.1.2.2 5,17-dioxo-A-nor-3,5-secoandrostan-3-oic acid (2)
To a solution of 1 (286.4 mg, 1 mmol) in isopropanol (6 mL) was added a heated solution of sodium carbonate (170 mg, 1.6 mmol) in water (1.5 mL). The mixture was brought to reflux and a solution of sodium periodate (1.79 g, 8.3 mmol) and potassium permanganate (23.3 mg, 0.15 mmol) in warm water (3 mL) was added dropwise over a period of 1 h and reflux was maintained for more 2 h. Then, the reaction was cooled to 30 °C and the solids were removed by filtration using Celite and washed with water. The filtrate was concentrated under reduced pressure to remove the organic solvent. The aqueous residue was cooled and acidified with concentrated hydrochloric acid until precipitate formation. Then, the product was extracted with ethyl acetate (3 × 40 mL), washed with brine, and dried with anhydrous sodium sulfate. After removal of the solvent under reduced pressure product 2 was obtained as a pallid yellow solid (265.6 mg, 83%); mp 114–115 °C; IR (cm−1): 3162, 2938, 2858, 1732, 1697.H NMR (CDCl3, 400 MHz) δ: 1.08 (3H, s, 19-CH3), 0.87 (3H, s, 18-CH3). 13C NMR (CDCl3, 100 MHz) δ: 220.46, 214.26, 179.21, 50.70, 50.47 47.98, 47,68 37.72, 35.73, 34.44, 30.98, 29.95, 29.23, 29.12, 21.84, 20.76, 20.43, 13.79.
4.1.2.3 4-azaandrost-5-ene-3,17-dione (3)
A mixture of 2 (245.1 mg, 0.8 mmol) and ammonium acetate (379.2 mg, 4.9 mmol) in glacial acetic acid (7 mL) was heated under reflux for 4 h. At the end of the reaction, the mixture was cooled and water (75 mL) was added. Then, the product was extracted with DCM (3 × 80 mL). The organic phase was washed with brine and dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure to give product 3 as a dark orange solid (232.1 mg, 98%); mp 246–249 °C; IR (cm−1): 3096, 2945, 2875, 1727, 1693, 1670, 860.1H NMR (CDCl3, 400 MHz) δ: 7.92 (1H, s, NH), 4.87 (1H, s, 6-H), 1.10 (3H, s, 19-CH3), 0.89 (3H, s, 18-CH3). 13C NMR (CDCl3, 100 MHz) δ: 219.44, 168.57, 139.05, 101.86, 50.50, 47.07, 46.60, 34.75, 33.34, 30.43, 30.28, 30.15, 27.61, 27.32, 20.79, 19.26, 17.75, 12.62.
4.1.3 General procedure for the preparation of 16E-arylidene-4-azaandrost-5-ene-3,17-dione derivatives by aldol condensation (4a–g)
To an ethanolic solution of compound 3 (57.5 mg, 0.2 mmol) and aldehyde (0.24 mmol) was added an aqueous solution of potassium hydroxide (60 μL, 50% m/m) and the reaction was stirred for 12–24 h at room temperature. The reaction mixture was worked up first by adding water to induce precipitation (15 mL) and then filtering and washing with water to give the 4-azaandrostene derivatives 4a–g.
4.1.3.1 16-(Phenylmethylidene)-4-azaandrost-5-ene-3,17-dione (4a)
Beige powder (41.9 mg, 56%); mp 278–280 °C; IR (cm−1): 3203, 3066, 2942, 2865, 1716, 1667, 1631, 973, 855, 772, 693.1H NMR (CDCl3, 400 MHz) δ: 7.90 (1H, s, NH), 7.47 (2H, d, J = 7.2 Hz, HAr 7.34 (4H, dt, J = 15.4, 9.6 Hz, HAr+vin), 4.83 (1H, s, 6-H), 1.10 (3H, s, 18-CH3), 0.94 (3H, s, 19-CH3). 13C NMR (CDCl3, 101 MHz) δ: 209.14, 169.58, 140.23, 135.55, 133.43, 130.33 (2C), 129.35, 128.72 (2C), 102.58, 49.66, 48.17, 47.40, 34.43, 31.42, 31.28, 30.97, 29.23, 28.78, 28.39, 20.32, 18.83, 14.33. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H30NO2 376.2271; Found 376.2273.
4.1.3.2 16-[(4-Methylphenyl)methylidene]-4-azaandrost-5-ene-3,17-dione (4b)
Beige powder (60.1 mg, 78%); mp 290–292; IR (cm−1): 3208, 3065, 2942, 2865, 1716, 1667, 1630, 984, 833, 771.1H NMR (CDCl3, 400 MHz) δ: 7.79 (1H, s, NH), 7.36 (3H, s, HAr) 7.17 (3H, d, J = 6.7 Hz, HAr+vin), 4.83 (1H, s, 6-H), 2.32 (3H, s, CH3), 1.09 (3H, s, 18-CH3), 0.93 (3H, s, 19-CH3). 13C NMR (CDCl3, 101 MHz) δ: 209.27, 169.54, 140.22, 139.75, 134.57, 133.49, 132.69, 130.38 (2C), 129.49 (2C), 102.60, 49.70, 48.18, 47.36, 34.45, 31.42, 31.29, 30.97, 29.25, 28.79, 28.39, 21.49, 20.33, 18.83, 14.36. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H32NO2 390.2428; Found 390.2432.
4.1.3.3 16-[(4-Nitrophenyl)methylidene]-4-azaandrost-5-ene-3,17-dione (4c)
Orange powder (61.5 mg, 75%); mp 275–277; IR (cm−1): 3188, 3057, 2939, 2860, 1718, 1668, 1634, 1596, 1515, 1341, 832, 772.1H NMR (CDCl3, 400 MHz) δ: 8.21 (2H, d, J = 8.7 Hz, HAr) 7.86 (1H, s, NH), 7.60 (2H, d, J = 8.8 Hz, HAr), 7.40 (1H, s, Hvin) 4.84 (1H, s, 6-H), 1.10 (3H, s, 18-CH3), 0.96 (3H, s, 19-CH3). 13C NMR (CDCl3, 101 MHz) δ: 208.24, 169.52, 147.58, 141.82, 140.27, 139.51, 130.67 (2C), 130.54 (2C), 123.92, 102.33, 49.40, 48.12, 47.54, 34.44, 31.41, 31.21, 30.97, 29.29, 28.76, 28.36, 20.27, 18.84, 14.26. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H29N2O4 421.2122; Found 421.2122.
4.1.3.4 16-[(furan-2-yl)methylidene]-4-azaandrost-5-ene-3,17-dione (4d)
Pallid yellow powder (55.2 mg, 66%); mp 290–291 °C; IR (cm−1): 3195, 3089, 2944, 2865, 1712, 1680, 1624, 1017, 833.1H NMR (CDCl3, 400 MHz) δ: 7.77 (1H, s, NH), 7.59 (1H, s, HAr), 7.29 (1H, s, Hvin), 6.68 (1H, s, HAr), 6.54 (1H, s, HAr), 4.92 (1H, s, 6-H), 1.18 (3H, s, 18-CH3), 0.99 (3H, s, 19-CH3). 13C NMR (CDCl3, 101 MHz) δ: 209.12, 175.92, 152.15, 144.84, 140.21, 133.02, 119.85, 115.87, 112.39, 102.61, 49.25, 48.21, 47.53, 34.47, 33.53, 31.46, 31.24, 30.95, 28.82, 28.52, 20.34, 18.82, 14.43. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H28NO3 366.2064; Found 366.2052.
4.1.3.5 16-[(5-methylfuran-2-yl)methylidene]-4-azaandrost-5-ene-3,17-dione (4e)
Yellow powder (67.5 mg, 85%); mp 196–198 °C; IR (cm−1): 3272, 2961, 1707, 1656, 1620, 1574, 1093, 796.1H NMR (CDCl3, 400 MHz) δ: 7.69 (1H, s, NH), 7.10 (1H, s, Hvin), 6.51 (1H, d, J = 3.5 Hz, HAr), 6.06 (1H, d, J = 3.1 Hz, HAr), 4.84 (1H, s, 6-H), 2.31 (3H, s, CH3), 1.09 (3H, s, 18-CH3), 0.90 (3H, s, 19-CH3). 13C NMR (CDCl3, 101 MHz) δ: 199.62, 169.72, 155.58, 140.71, 140.04, 136.61, 133.31, 131.27, 117.58, 109.02, 49.36, 48.20, 47.49, 34.47, 31.69, 31.26, 30.92, 28.81, 28.28, 20.34, 18.81, 14.47, 14.11. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H30NO3 380.2220; Found 380.2215.
4.1.3.6 16-[(pyridin-3-yl)methylidene]-4-azaandrost-5-ene-3,17-dione (4f)
Beige solid (59.0 mg, 79%); mp 266–268 °C; IR (cm−1): 3236, 2942, 1715, 1667, 1636, 1587, 1480, 826.1H NMR (CDCl3, 400 MHz) δ: 8.73 (1H, s, HAr), 8.52 (1H, s, HAr), 7.84–7.72 (2H, m, NH + HAr), 7.32 (2H, dd, J = 14.7, 9.5 Hz, HAr+vin), 4.83 (1H, s, 6-H), 1.10 (3H, s, 18-CH3), 0.95 (3H, s, 19-CH3). 13C NMR (CDCl3, 101 MHz) δ: 208.38, 169.45, 151.08, 149.70, 140.26, 137.80, 136.89, 131.43, 129.60, 123.62, 102.37, 49.52, 48.13, 47.46, 34.45, 31.42, 31.22, 30.96, 29.26, 28.75, 28.38, 20.29, 18.83, 14.29. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H29N2O2 377.2224; Found 377.2225.
4.1.3.7 16-[(thiophen-2-yl)methylidene]-4-azaandrost-5-ene-3,17-dione (4g)
Dark orange powder (64.2 mg, 84%); mp 297–298 °C; IR (cm−1): 3195, 3059, 2942, 2864, 1710, 1664, 1615, 1096, 832.1H NMR (CDCl3, 400 MHz) δ: 7.97 (1H, s, NH), 7.57 (1H, s, HAr), 7.45 (1H, d, J = 19.7 Hz, HAr), 7.27 (1H, s, Hvin), 7.09–7.04 (1H, m, HAr), 4.86 (1H, s, 6H), 1.09 (3H, s, 18-CH3), 0.91 (3H, s, 19-CH3). 13C NMR (CDCl3, 101 MHz) δ: 208.89.69.60, 140.24, 139.82, 133.25, 132.47, 129.82, 127.95, 126.08, 102.62, 49.41, 48.21, 47.78, 34.44, 31.43, 31.25, 30.93, 28.94, 28.79, 28.39, 20.32, 18.83, 14.50. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H28NO2S 382.1835; Found 382.1836.
4.2 Biology
4.2.1 Cell cultures
LNCaP, PC-3, T47-D, and NHDF cells were obtained from American Type Culture Collection (Manassas, VA) and were cultured in 75 cm2 culture flasks at 37 °C in a humidified air incubator with 5% CO2. NHDF cells are healthy fibroblasts of the human adult dermis. LNCaP, PC-3, and T47-D cells, which were used in passages 20–26, 25–28, and 10–13, respectively, were cultured in RPMI 1640 medium (Sigma–Aldrich, Inc., St. Louis) with 10% fetal bovine serum (Sigma–Aldrich, Inc.) and 1% of the antibiotic mixture of 10,000 U/mL penicillin G and 100 mg/mL of streptomycin (Sp, Sigma–Aldrich, Inc.). Finally, NHDF cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, and 1% of the antibiotic/antimycotic (10,000 U/mL penicillin G, 100 mg/mL streptomycin, and 25 μg/mL amfothericin B) (Ab; Sigma–Aldrich, Inc.), and these cells were used in passages 10–12. For all cell types, the medium was renewed every 2–3 days until cells reach nearly the confluence state. When cells reach approximately 90–95% confluence, they were detached gently by trypsinization (trypsin–EDTA solution, 0.125 g/L of trypsin and 0.02 g/L of EDTA). Before each experiment, viable cells were counted, in a Neubauer chamber by a trypan blue exclusion assay and adequately diluted in the appropriate complete cell culture medium.
4.2.2 Preparation of compounds solutions
All compounds were dissolved in dimethyl sulfoxide (DMSO; Sigma–Aldrich, Inc.) at a concentration of 10 mM and stored at 4 °C. From the mother solutions, the various diluted solutions of the compounds in the study at different concentrations were prepared in a complete culture medium before each experiment. The maximum level of DMSO concentration in the studies was 1% and previous studies demonstrated that this concentration has no relevant effects on cell proliferation (data not shown).
4.2.3 MTT assay
As previously referred, after reaching a near confluence state, cells were trypsinized and counted by the trypan blue exclusion assay, and then 100 μL of cell suspension/well with an initial density of 2 × 104 cells/mL was seeded in 96-well culture plates (Nunc, Apogent, Denmark) and left to adhere for 48 h. After the cell adherence, the medium was replaced by several solutions of the compounds in this study (30 μM for preliminary studies and 0.01, 0.1, 1, 10, 50, and 100 μM for concentration–response studies) in an appropriate medium for approximately 72 h. 5-FU and finasteride were used as positive controls and untreated cells were used as the negative control. Each experiment was performed in quadruplicate and independently repeated at least two times [43].
The in vitro antiproliferative effects were evaluated by the MTT assay (Sigma–Aldrich, Inc.). After the incubation period, the medium was removed and 100 μL of phosphate buffer saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 in deionized water and pH adjusted to 7.4) was used to wash the cells. Then 100 μL of the MTT solution (5 mg/mL) was prepared in the appropriate serum-free medium and was added to each well, followed by incubation for 4 h at 37 °C. Hereafter, the MTT-containing medium was removed and the formazan crystals were dissolved in DMSO. Then the absorbance was measured at 570 nm using a microplate spectrophotometer BIO-RAD xMark. Cell viability values were expressed as percentages relatively to the absorbance determined in the cells used as negative controls.
4.2.4 Flow cytometry assay
The analysis of cell viability was performed by flow cytometry after staining dead cells with PI (Invitrogen). Briefly, 1 mL of a cell suspension was seeded in 12-well culture plates (initial cell density of 5 × 104 cells/mL of LNCaP cells for 24 h assay and 2 × 104 cells/mL of the same cell line for 72 h assay) in complete culture medium. After 48 h the cells were treated with finasteride and 5-FU, as positive controls, and compound 4b at a concentration of 50 μM. Untreated cells were used as a negative control.
At the pretended time point, the supernatant was collected and pooled with the cells harvested by trypsin treatment (each well was also washed with 400 μL of PBS before trypsin treatment). The resulting cell suspension was kept on ice and pelleted by centrifugation and resuspended with 400 μL of complete medium. Afterward, 5 μL of PI (1 mg/mL) was transferred to a FACS tube and 395 μL of the cell suspension was also added to the same tube. Twenty thousand events (very small events excluded) were acquired using a FACSCalibur flow cytometer, using the flow cytometry standard (FCS), side-scattered light (SSC), and FL3 (PI) channels. Acquisition and analysis were performed with CellQuest Pro software (v. 5.1). Briefly, a region was created on the SSC/FCS contour plot to exclude events of very small size and complexity (considered unrelevant debris). Then, in the FCS/FL3 contour plot gated on the previous region, three regions were created, first corresponding to viable cells, R1, second to dead cells, R2, and third corresponding to an intermediate subpopulation of cells, R3, which may include both large debris resulting from apoptotic death (with very low DNA content) and cells with partial permeability to PI. The percentages of each region were calculated for a total of R1 + R2 + R3. The experiment was performed in two dependent days, each in duplicate or triplicate wells.
4.2.5 Data analysis
The data are expressed as a mean ± standard deviation (SD) in MTT and flow cytometry assays. Differences between groups were evaluated by means of the Student t test and one-way analysis of variance analysis and were considered statistically significant when <0.05. The determination of IC50 was done by sigmoidal fitting analysis considering a 95% confidence interval. All data shown are representative of at least two independent experiments.
4.3 Molecular docking studies
4.3.1 Preparation of proteins
The three-dimensional structural coordinates for 5BR (PDB code, 3G1R), ERα (PDB code, 1A52), AR (PDB code, 2AMA), CYP17A1 (PDB code, 3RUK), and aromatase (PDB code, 3EQM) were obtained from PDB (www.rcsb.org). The coordinates of the ligands cocrystallized and water molecules were selected using the software Chimera (v. 1.10.1) and histidine charges were defined to match a physiological environment, and the final structures were saved in the PDB format. Then nonpolar hydrogen atoms were merged in AutoDockTools (v. 1.5.6) from The Scripps Research Institute [44]. Kollman and Gasteiger partial charges were added in the same software. Finally, the prepared structures were converted from the PDB format to PDBQT for posterior utilization in the docking study.
4.3.2 Preparation of ligands
All ligands were constructed using Chem3D (v. 12.0) software (by Cambridge ChemBioOffice 2010). Energy minimization and geometry optimization (MMFF94 force field, 500 steps of conjugate gradient energy minimization followed by 500 steps of steepest descent energy minimization with a convergence setting of 10 × 10−7) were performed with Avogadro (v. 1.0.1), and the final structures were saved as PDB file format. Then, the ligands were completely prepared choosing torsions and the structures were converted from PDB format to PDBQT in AutoDockTools.
4.3.3 Grid map calculations
AutoDock grid maps were calculated for each macromolecule using AutoGrid4, based on the active site coordinates of each protein crystal structure. The size of all grid boxes was 40 × 40 × 40 with 0.375 Å of spacing. Maps were calculated for each atom type in each ligand along with an electrostatic and desolvation map using a dielectric value of −0.1465.
4.3.4 Molecular docking simulations
Molecular docking simulations were conducted using the Lamarckian genetic algorithm and empirical free energy scoring function [45]. The maximum number of energy evaluations was 2,500,000 and the GA population size was 150. A total of 15 hybrid global-local Lamarkian search (GA-LS) runs were performed for each simulation. The results of molecular docking were visualized in PyMol program (PyMol Molecular Graphics System, v. 1.3, Schrödinger, LLC – www.pymol.org), built for educational use. All docking simulations performed to validate the method, using the ligands present in crystal structures, were able to reproduce the ligand–protein interaction geometries. For the docking process to be considered successful, the RMSD value between ligand conformations (docked ligand and crystallized ligand) was <2.0 Å.
Acknowledgments
This work is supported by FEDER funds through the POCI – COMPETE 2020 – Operational Programme Competitiveness and Internationalization in Axis I – Strengthening Research, Technological Development and Innovation (Project No. 007491) and National Funds by the FCT – Foundation for Science and Technology (Project UID/Multi/00709). VB also acknowledges the grants BID/ICI-FC/Santander Universidades-UBI/2016 and SFRH/BD/131059/2017 (FCT).