


 CC-BY 4.0
CC-BY 4.0
1. Introduction
The Tsuji–Trost allylation is a pivotal reaction in synthesis because of its operational simplicity and its great potential for applications in total synthesis [1, 2, 3]. As part of the development of this chemistry, the design and the synthesis of simple ligands remain of primary importance [4, 5]. In terms of sustainable chemistry, using natural products as precursors for the design of new ligands is particularly judicious due to their low cost and their availability in large quantities. The presence of functionalities such as ketones and carbon–carbon double bonds are suitable to introduce N-coordination centres. In contrast to phosphines, nitrogen chelates would be preferable owing to their higher stability towards oxidation processes. Moreover, several starting building blocks are chiral and thus additionally open the possibility to access asymmetric transformations [6]. In that context, the family of natural terpenes is a convenient source to produce ligands [7, 8]. Among the large variety of ligands issued from terpenes, 𝛼-amino-oximes ligands have been synthesized and used with different metals [9] such as Pd [10], Zn and Pt [11], Cu [12], Co [13], or Ni [14] in several fields, from biochemistry and pharmacology applications [15] to organometallic catalysis and asymmetric synthesis. As an example, they have already been applied in catalytic reactions such as homocoupling of amines [16] and epoxidation of olefins [17]. In our group, a new family of optically pure bifunctional 𝛼-amino-oxime ligands based on (R)-limonene has been synthesized and used as chiral inducers for enantioselective hydrogen transfer reactions on various ketones in the presence of ruthenium catalysts [18]. Moreover, one of them has recently been used in combination with palladium precursor leading to an enantiopure palladium complex showing medium anticancer activities [19].
Herein, we report on the use of 𝛼-amino-oxime ligands to stabilize palladium-based complexes and the use of such precursors to promote allylic alkylation reactions. To our knowledge, despite the impressive number of various ligands [20] used in this reaction, including some issued from the chiral pool such as monoterpenes [21] or sugars [22], no 𝛼-amino-oxime ligand has been described for the allylic substitution.
As part of our ongoing research towards the development and their applications as new catalysts, we report here the synthesis of new Pd (II) complexes bearing enantiomerically pure 𝛼-amino-oxime ligands derived from (R)-limonene. The molecular structures were determined by NMR and X-ray analyses and their catalytic properties were evaluated in the model allylic alkylation of dimethylmalonate with allylacetate and more generally of various substrates.
2. Results and discussion
2.1. Synthesis and characterization of palladium compounds
The synthesis of the optically pure (1S,4R)-amino-oximes from (R)-limonene proceeds via the nitrosochloride intermediate formation following the addition of an amine [18]. Three different amines like aniline, benzylamine and 2-picolylamine were used leading to three optically pure bidentate 𝛼-amino-oximes ligands L1–L3, respectively. Royo proposed a synthesis of complexes from precursors K2PdCl2 or Pd(cod)Cl2 (cod = cycloocta-1,5-diene) and L3 ligand leading to the cationic complex {Pd(L3)Cl}+,Cl− in which the picolyl-amino-oxime with the three nitrogen atoms acts as a tridentate ligand [19]. In our case, the reactions of the precursor PdCl2(CH3CN)2 with the amino-oximes L1 and L2 afforded the clean formation of the new chiral and neutral complexes C1 and C2 of general formula PdCl2(L) (L = L1 or L2) as orange powder and with yields of 87 and 93% respectively. In the case of the tridentate ligand L3, the cationic complex C3 was obtained as expected (Scheme 1). The new palladium complexes C1 and C2 were fully characterized by 1H and 13C NMR analyses. As described in the literature, the configurations of the ligands in those compounds were assumed to be (2S,5R) which is confirmed by X-ray structures.
Synthesis of enantiomerically pure palladium complexes.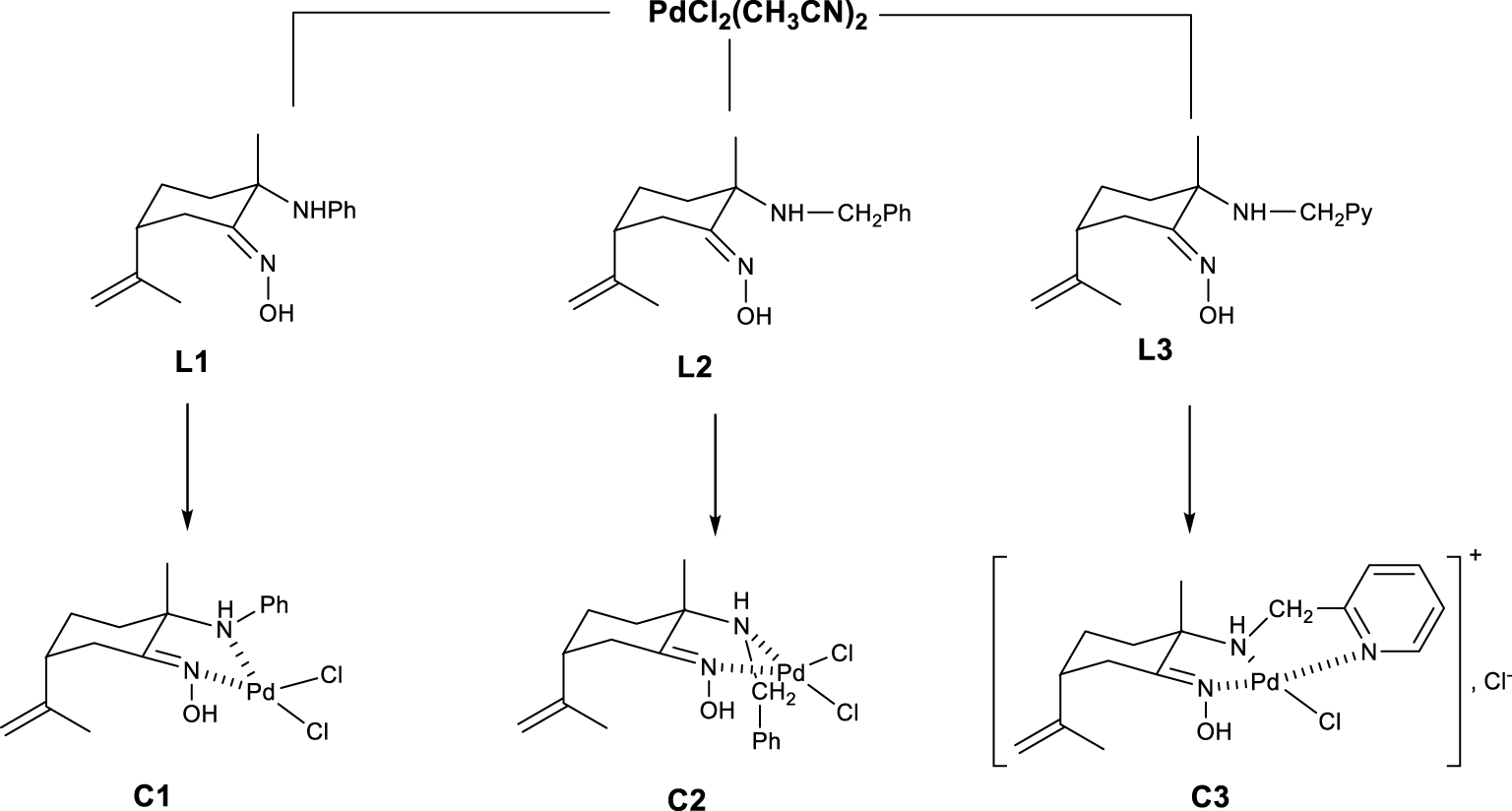
NMR spectroscopic data revealed the presence of the complexes C1 and C3 in CDCl3 as single species while, in the case of the complex C2, two diastereoisomers were observed in a ratio of about 75:25. The five-membered chelate rings PdN2C2 were formed via coordination of the nitrogen atoms from the amino and the amino-oxime groups. No hydrogen abstraction from these groups were observed, the corresponding proton signals being retained in the 1H NMR spectra of the complexes (see experiment part).
Thus, the NMR data comparison between the ligand L1 and its complex C1 showed noticeable changes in the close environment of the coordinated nitrogen atoms. The signals of NH protons were strongly downfield shifted from 3.57 to 8.10 ppm and the N–OH signal shifted from 8.16 to 10.00 ppm. Concerning the 13C NMR spectra, a similar behaviour is observed around the carbon atoms linked to the N atoms (𝛿Cq‐NOH 164.79 to 171.37 ppm and 𝛿Cq‐NH 56.66 to 73.16 ppm from the free ligand to the complex). These values are in total agreement with data collected for L2PdCl2 complexes (L = 𝛼-(o-anisidino)caranone and 𝛼-(o-anisidino)pinanone) [10]. In the case of the ligand L3 and the corresponding complex C3, as described by Royo [19], similar observations could be made: the resonance of the proton of the N–OH shifted from 9.30 ppm in the ligand to 8.62 ppm for the complex. Similarly, signals due to carbon Cq-NOH, shifted from 161.51 ppm for the ligand L3 to 181.64 ppm in C3 and the signal of the Cq-NH shifted from 56.54 to 68.33 ppm. These significant downfield shifts indicated clearly the coordination of both amino and oxime nitrogen atoms to palladium.
Unexpectedly, analysis of the 1H NMR spectrum of the complex C2 showed two broad signals at 𝛿 9.96 and 9.80 ppm assigned to the OH protons of two diastereoisomers C2 and C ′2 present in solution. Kokina observed such results with complexes of PdCl2 coordinated by amino-oximes ligand issued from carene [10]. He proposed that the two diastereoisomers differ by the configuration of the N atom of the NH group. A similar behaviour could be envisioned in our case. The major product would show a trans-arrangement of the H atom and the methyl group in respect to the chelate PdN2C2, while the minor product would be characterized by a cis-arrangement. The chemical shifts were slightly shifted in comparison to the free ligand L2 for which the resonance was observed at 9.23 ppm. In the same way, the difference between the ligand L2 and the corresponding complex C2 in the 13C NMR spectrum was noticeable with a shift from 162.85 to 170.45 ppm for the Cq-NOH and the signals assigned to Cq-NH from 56.52 to 71.73 ppm.
The structure of the new palladium-amino-oxime complexes C1 and C2 were unequivocally elucidated by single-crystal X-ray diffraction analysis. Suitable single crystals were grown by slow diffusion at room temperature of diethyl ether into a dichloromethane solution of the mononuclear palladium complexes C1 and C2. Despite numerous trials, we were unable to crystallize the C3 complex.
Molecular structure of the palladium complex C1 shown with 50% probability ellipsoids.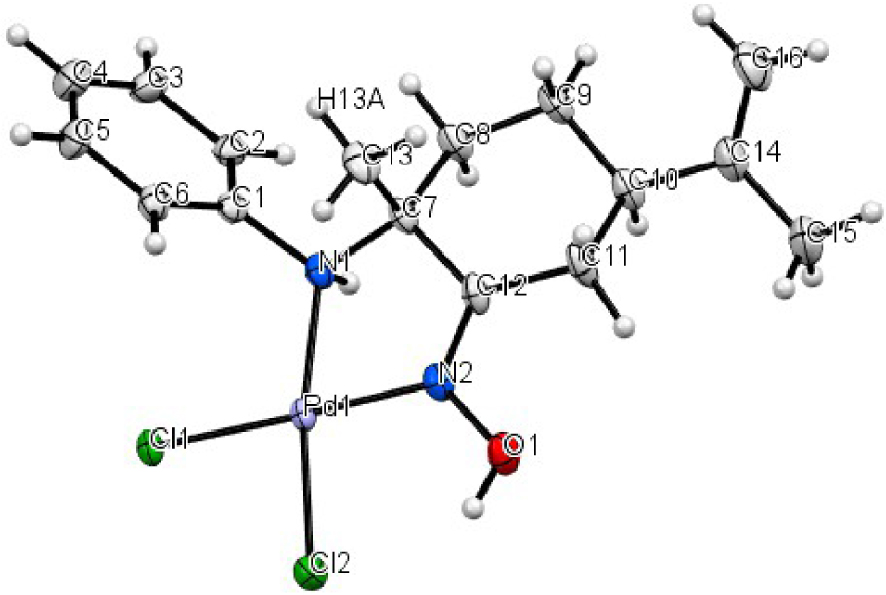
ORTEP plots of the two complexes C1 and C2 are shown in Figures 1 and 2 and some relevant structural parameters are collected in Table 1. The diffraction studies showed that the complex C1 crystallized in a trigonal space group and the coordination geometry around the metal is a square planar with N(1)–Pd–N(2) and Cl(1)–Pd–Cl(2) angles of 80.10° and 91.80°, together with angles Cl(2)–Pd–N(2), and N(1)–Pd–Cl(1) of 91.4° and 96.5° respectively, indicating that the geometry of the square planar is slightly distorted (Figure 1). The two Cl atoms are in a cis-position with Pd–Cl distances of 2.288 and 2.307 Å which are comparable with those reported in the literature.
The NMR of the complex C2 showed the formation of two diastereoisomers. Only one of them was isolated in the crystalline state. The crystallization is formed in the P212121 chiral space group. The coordination geometry of the complex C2 was a distorted square resulting from the ring closure of a five-membered chelating cycle PdN2C2 with a 80.60° N(1)–Pd–N(2) angle. The lengths of Pd–N bonds are respectively 1.990 Å and 2.044 Å. Almost the same bond lengths as those observed in the complex C1 were found.
Molecular structure of the palladium complex C2 shown with 50% probability ellipsoids.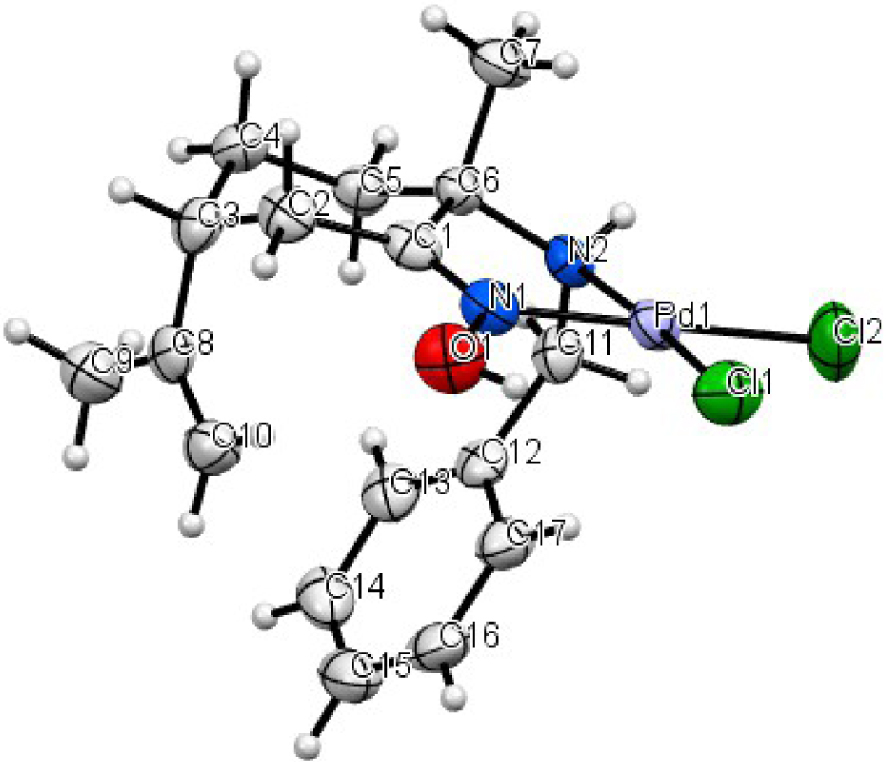
Selected bond lengths (Å) and angles (deg) for C1 and C2
| Complex | C1 | C2 |
|---|---|---|
| Pd–Cl(1) | 2.288(1) | 2.3187(5) |
| Pd–Cl(2) | 2.307(8) | 2.2886(5) |
| Pd–N(1) | 2.071(3) | 1.990(1) |
| Pd–N(2) | 1.981(4) | 2.044(1) |
| Cl–Pd–Cl | 91.80(4) | 94.56(2) |
| N(1)–Pd–N(2) | 80.10(1) | 80.60(6) |
2.2. Catalytic studies
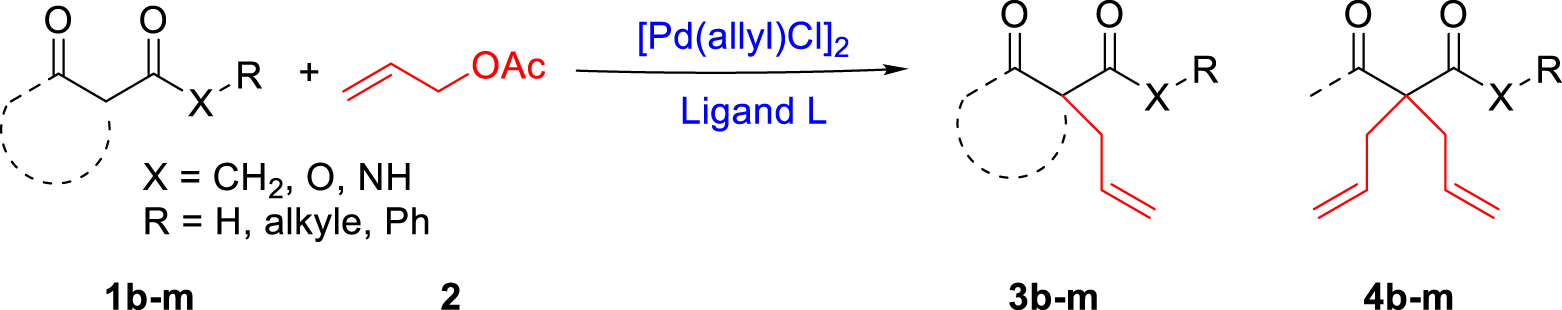
The three 𝛼-amino-oximes were then applied in the palladium-allylic alkylation. We used the in situ technique to evaluate the catalytic performance of the ligands with [PdCl(allyl)]2 as catalytic precursor. The catalysts were formed by mixing [PdCl(allyl)]2 with two equivalents of the desired 𝛼-amino-oxime in the chosen solvent.
The optimization of the reaction conditions was first carried out with dimethylmalonate 1a and allyl acetate 2 as substrates in the presence of the Pd/L3 catalytic system (Table 2). The reaction led to the formation of both the monoallylated malonate compound as major product 3a and the bis-allylated compound 4a as a minor product.
Optimization of the reaction palladium-catalysed allylation of dimethylmalonate using chiral ligandsa

| Entry | L | Solvent (mL) | T (°C) | Base (mol%) | Conv.b (%) | 3ab (%) | 4ab (%) |
|---|---|---|---|---|---|---|---|
| 1 | L3 | THF (0.5) | 80 | Cs2CO3(10) | 4 | 4 | — |
| 2 | L3 | THF (0.5) | 60 | Cs2CO3(10) | 19 | 15 | 4 |
| 3 | L3 | MeOH (0.5) | 60 | Cs2CO3(10) | 29 | 13 | 16 |
| 4 | L3 | Toluene (0.5) | 60 | Cs2CO3(10) | 23 | 15 | 8 |
| 5 | L3 | Dioxane (0.5) | 60 | Cs2CO3(10) | 17 | 12 | 5 |
| 6 | L3 | DMF (0.5) | 60 | Cs2CO3(10) | 28 | 17 | 11 |
| 7 | L3 | THF (0.5) | 60 | Cs2CO3(20) | 31 | 23 | 8 |
| 8 | L3 | THF (0.5) | 60 | Cs2CO3 (30) | 39 | 30 | 9 |
| 9 | L3 | THF (0.5) | 60 | Cs2CO3 (50) | 60 | 50 | 10 |
| 10 | L3 | THF (0.5) | 60 | Cs2CO3(100) | 70 | 60 | 10 |
| 11 | L3 | THF (0.5) | 60 | NaOH (10) | 20 | 12 | 8 |
| 12 | L3 | THF (0.5) | 60 | K2CO3(100) | 53 | 38 | 15 |
| 13 | L3 | THF (1.0) | 60 | K2CO3(100) | 88 | 69 | 17 |
| 14 | L3 | THF (2.0) | 60 | K2CO3(100) | 76 | 64 | 12 |
| 15 | L2 | THF (2.0) | 60 | K2CO3(100) | 80 | 65 | 15 |
| 16 | L1 | THF (2.0) | 60 | K2CO3(100) | 86 | 70 | 16 |
| 17 | — | THF (2.0) | 60 | K2CO3(100) | 20 | 18 | 2 |
a Conditions: 1a (3.6 mmol), 2 (7.2 mmol), [Pd(allyl)Cl]2: 3 mol%, Ln = 6 mol%, 17 h.
b Determined by gas chromatography (GC).
The first reaction was carried out in the presence of Pd/L3 catalyst at 80 °C for 17 h with THF as solvent and Cs2CO3 as base in catalytic amount (entry 1). A very poor conversion was observed with only 4% of monoallylmalonate 3a. Decreasing the temperature to 60 °C allowed a slight increase of the conversion up to 19% with the mono derivative as major product despite the two equivalents of allylic acetate vs malonate (entry 2). This result could be due to a greater stability of the complex at 60 °C rather than 80 °C. Attempts were made to improve the conversion upon using other solvents like methanol, toluene, dioxane and DMF (entries 3–6). Conversions between 17% to 29% and 3a yields from 12% to 17% were observed. Nevertheless, the best result in terms of malonate conversion and selectivity into the monoallylated product 3a was obtained with THF as solvent (entry 2). At 60 °C and in the presence of THF as solvent, the amount of base was gradually increased to 100 mol% leading, as expected, to better conversions up to 70% with one equivalent of base related to the dimethylmalonate 1a (entries 2, 7–10). As Cs2CO3 is an expensive base, other bases like NaOH (entry 11) and K2CO3 (entry 12) were used leading to similar results. The result obtained with K2CO3 in 1 mL THF was interesting with a conversion increased to 88% and in that condition, the yield into 3a attained 69% (entry 13). In order to improve the conversion, the amount of solvent was again increased to 2 mL (entry 14). However, this addition did not give a better result. The ligands L1 and L2 were also evaluated as reaction promoters (entries 15 and 16). Close conversions and yields into the monoallylated product 3a were observed probably due to the structural similarity of the three amino-oximes. It is noteworthy that conversion is very poor (20%) in the absence of the amino-oxime ligand (entry 17).
The experimental conditions (ligand L3, T = 60 °C and 2 mL THF with 100% K2CO3) were extended to a series of racemic substrates with different functionalities like 𝛽-diketone, 𝛽-ketoesters and 𝛽-ketoamides (Table 3).
Substrates scope for allylation with allyl acetate 2a
| Entry | 1b–m | Conv. GC (%) | 3 GC yield (%) | 4 GC yield (%) | Isol. yield (%) |
|---|---|---|---|---|---|
| 1 | 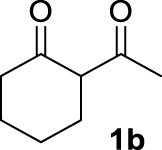 | 100 | 100 | — | 72 (3b) |
| 2 | 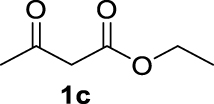 | 95 | 64 | 31 | 42 (3c) |
| 3 | 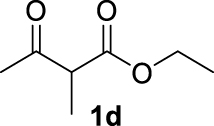 | 70 | 70 | — | 61 (3d) |
| 4 | 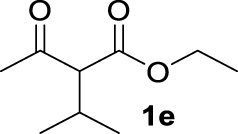 | 69 | 69 | — | 24 (3e) |
| 5 | 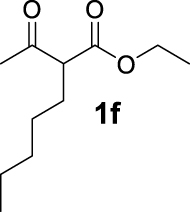 | 50 | 50 | — | 20 (3f) |
| 6 | 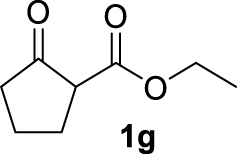 | 83 | 83 | — | 64 (3g) |
| 7 | 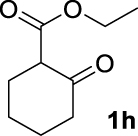 | 83 | 83 | — | 75 (3h) |
| 8 | 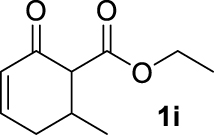 | 49 | 49 | — | 38 (3i) |
| 9 | 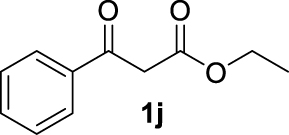 | 54 | 43 | 11 | 31 (3j) |
| 10 | 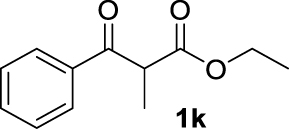 | 71 | 71 | — | 62 (3k) |
| 11 | 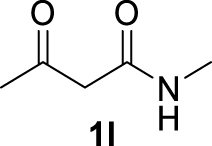 | 86 | 54 | 32 | — |
| 12 | 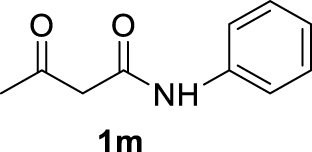 | 100 | 100 | 0 | — |
a Conditions: Substrate 1b–m (3.6 mmol), allylacetate 2 (7.2 mmol), [Pd(allyl)Cl]2: 3 mol%, L3: 6 mol%, K2CO3 (3.6 mmol), THF = 2 mL; 60 °C, 17 h.
The 𝛽-diketone 1b was allylated with 100% GC conversion and 72% of isolated yield into the monoallylated product 3b (entry 1, Table 3). The current allylation could be successfully applied to 𝛽-ketoesters (1c–k). In the case of ethyl-2-oxobutanoate 1c, a mixture of the mono (64%) and the di (31%) compounds was obtained (entry 2). The conversion was slightly lower than those obtained with the corresponding diketone 1a (Table 1, entry 14) due to the lower acidity of the proton of the methylene group in the ketoester compared to the diketone. Then 𝛽-ketoesters substituted in the methylene position (1d–i and 1k) were evaluated. As expected, these substrates could be converted into the monoallylated derivatives which have been isolated in various yields from 20% to 75% (entries 3–8 and 10). Ketoamides 1l and 1m proved to be good substrates for the allylation with this catalytic system leading to high conversions (86% and 100% respectively, entries 11 and 12). Moreover, the reaction was 100% chemoselective towards the monoallylated compound 3m in the case of N-phenyl-2-oxo butanamide 1m (entry 12). Nevertheless, we were unable to isolate the amides 3l–m. Unfortunately, whatever the catalytic system used, no enantioselectivity on allylmalonate derivatives 3b–m could be observed.
3. Conclusion
In summary, we have synthesized new palladium complexes with 𝛼-amino-oxime ligands based on (R)-limonene skeleton. X-ray diffraction studies as well as 1H and 13C NMR in solution were used to characterize the complexes both in solid state and solution. The ligands act as N–N chelates towards palladium. The catalytic performance of these complexes have been studied in the allylic C–H alkylation of the 𝛽-diester malonate, 𝛽-diketones, 𝛽-ketoesters and 𝛽-ketoamides with allyl acetate which led to interesting conversions but no enantioselectivity was observed in that reaction. Further investigations on the applications of amino-oximes in other asymmetric catalysis reactions are in progress.
4. Experimental
4.1. General methods
The reactions were performed under nitrogen atmosphere using standard Schlenk techniques. All reagents were commercial grade materials and were used without any further purification. Prior to use, the solvents were dried, distilled under N2 and deoxygenated in the vacuum line.
The 1H and 13C NMR spectra were recorded on a Bruker AC 300 spectrometer and referenced to TMS. PdCl2 was purchased from Strem chemicals (⩾99.9%) and PdCl2(CH3CN)2 was prepared as described in literature [23].
The ligands (2S,5R)-2-methyl-2-(phenylamino)-5-(prop-1-en-2-yl)cyclohexan-1-one oxime L1, (2S,5R)-2-(benzylamino)-2-methyl-5-(prop-1-en-2-yl)cyclohexan-1-one oxime L2 and (2S,5R)-2-methyl-5-(prop-1-en-2-yl)-2-((pyridin-2-ylmethyl)amino)cyclohexan-1-one oxime L3 were prepared according to published methods (Figure 3) [18].
𝛼-amino-oxime ligands L1–L3 from (R)-limonene.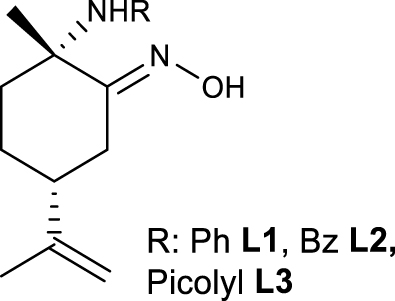
Synthesis of the PdCl2Ln (C1–C3 complexes).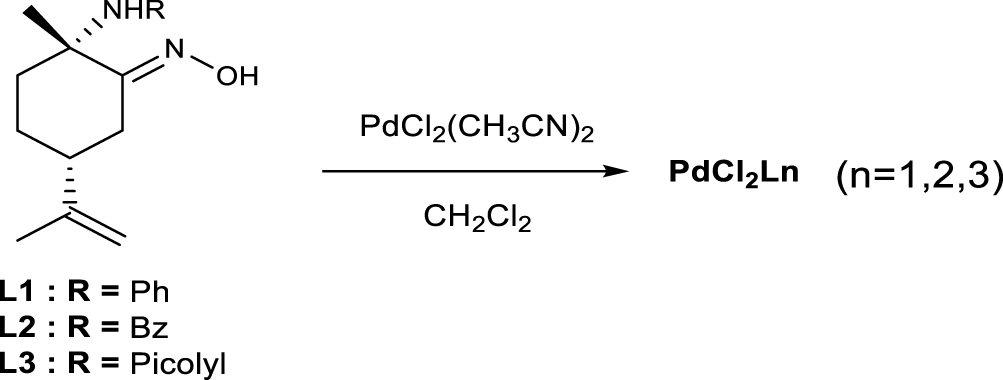
4.2. Synthesis of Pd complexes PdCl2(L) (L = L1 or L2 or L3)
A mixture of PdCl2(CH3CN)2 (0.54 mmol) and the ligand L1–L3 (0.54 mmol) in dichloromethane (60 mL) was stirred for 24 hours at room temperature. The evaporation of the solvent under vacuum led to an orange precipitate. The precipitate was washed with diethyl ether and filtered yielding the desired compounds (Figure 4).
For C15H22Cl2N2OPd (C1): Yield = 87%
For C16H24Cl2N2OPd (C2): Yield = 93%
For C15H23Cl2N3OPd (C3): Yield = 91%
4.3. NMR characterization of Ligands L1–L3 and complexes C1–C3
1H and 13C NMR spectra of ligand L1 and complex C1 in CDCl3
| Atom or group | L1 𝛿H (ppm) | C1 𝛿H (ppm) |
|---|---|---|
| =N–OH | 8.16, s, 1H | 10, s, 1H |
| N–H | 3.57, s, 1H | 8.10, s, 1H |
| –Ph | 6.69–7.16, m, 5H | 7.07–7.96, m, 5H |
| C =CH2 | 4.76, d, 2H | 4.78, d, 2H |
| C*H | 3.29–3.33, dt, 1H | 3.38–3.36, s, 1H |
| CH–CH2–CH2 | 2.22–2.02, q, 2H | 2.48–2.56 m, 2H |
| 2 CH2 | 2.01–1.53, m, 4H | 1.77–2.16 m, 4H |
| CH3 | 1.75, s, 3H | 1.73, s, 3H |
| CH3 | 1.47, s, 3H | 1.37, s, 3H |
| Atom or group | L1 𝛿C (ppm) | C1 𝛿C (ppm) |
| Cq =N–OH | 164.79 | 171.37 |
| Cq =CH2 | 148.23 | 146.28 |
| Cq(C6H5) | 146.52 | 138.56 |
| C6H5 | 129.18 | 131.08 |
| 118.26 | 127.89 | |
| 127.62 | ||
| 114.98 | 125.45 | |
| CH2= | 109.61 | 110.45 |
| Cq–NH | 56.66 | 73.16 |
| C*H– | 45.55 | 46.01 |
| CH2–(C*–NH) | 43.21 | 41.22 |
| CH2–C =NOH | 26.35 | 35.71 |
| CH2–CH–Cq | 25.45 | 30.68 |
| CH3–(C =CH2) | 22.81 | 27.40 |
| CH3–(C–NH) | 20.70 | 24.14 |
1H and 13C NMR spectra of ligand L2 and complexes C2 and C2′ in CDCl3
| Atom or group | L2 𝛿H (ppm) | C2 𝛿H (ppm) | C2′ 𝛿H (ppm) |
|---|---|---|---|
| =N–OH | 9.23, s, 1H | 9.96, s, 1H | 9.80, s, 1H |
| N–H | 2.6, s, 1H | — | — |
| –Ph | 7.30–7.13, m, 5H | 7.59–7.22, m, 5H | 7.59–7.22, m, 5H |
| C=CH2 | 4.72, s, 2H | 5.07, s, 1H | 5.55, s, 1H |
| 5.02, s, 1H | 5.58, s, 1H | ||
| CH2–Ph | 3.67, d, 1H | 5.03–4.54, m, 1H | 5.03–4.54, m, 1H |
| 3.38, d,1H | 3.85, 1H, dd | 4.05, dd, 1H | |
| C*H | 3.30, 1H, dt | 3.25, m, 1H | 3.35, m, 1H |
| 2 CH2 | 1.73-1.13, m, 4H | 1.42–0.95, m, 4H | 1.42–0.95, m, 4H |
| CH2–(C=NOH) | 1.55, d, 2H | 2.31, m, 1H | 2.44, m, 1H |
| 2.32, m, 1H | 2.22, m, 1H | ||
| CH3–(C=CH2) | 1.71, s, 3H | 1.80, s, 3H | 1.96, s, 3H |
| CH3–(C–NH) | 1.30, s, 3H | 1.53, s, 3H | 1.66, s, 3H |
| Atom or group | L2 𝛿C (ppm) | C2 𝛿C (ppm) | |
| Cq =N–OH | 162.85 | 170.45 | |
| Cq =CH2 | 148.53 | 145.19 | |
| Cq (C6H5) | 141 | 143.61 | |
| C6H5 | 128.48 | 129.51 | |
| 128.18 | 129.20 | ||
| 126.83 | 128.85 | ||
| 128.63 | |||
| 128.42 | |||
| CH2= | 109.54 | 112.75 | |
| Cq–NH | 56.52 | 71.73 | |
| C*H | 44.82 | 37.55 | |
| CH2–(C6H5) | 46.76 | 52.68 | |
| CH2–(C*–NH) | 40.57 | 34.48 | |
| CH2–C–NOH | 26.04 | 29.20 | |
| CH2–CH–Cq | 25.28 | 24.61 | |
| CH3–(C =CH2) | 23.27 | 23.13 | |
| CH3–(C–NH) | 20.81 | 21.06 | |
1H and 13C NMR spectra of ligand L3 and complex C3 in DMSO
| Atom or group | L3 𝛿H (ppm) | C3 𝛿H (ppm) |
|---|---|---|
| =N–OH | 9.30, s, 1H | 8.62, s, 1H |
| N-H | 2.42, s, 1H | — |
| –Py | 8.47, d, 1H | 8.42, d, 1H |
| 7.75, t, 1H | 8.05, d, 1H | |
| 7.46, d, 1H | 7.50, m, 2H | |
| 7.22, t, 1H | ||
| CH2–Py | 3.72, d, 1H | 4.90, s, 1H |
| 3.45, d, 1H | 4.51, s, 1H | |
| =CH2 | 4.74, d, 2H | 4.74–4.84, dd, 2H |
| 2 CH2 | 1.77–1.99, m, 4H | 2.50, m, 3H |
| 3.23–3.28, 1H, m | ||
| CH2 | 1.49, d, 2H | 1.73, m, 2H |
| C*H | 3.13, m, 1H | 3.00, m, 1H |
| CH3 | 1.72, s, 3H | 1.85, s, 3H |
| CH3 | 1.19, s, 3H | 1.64, s, 3H |
| Atom or group | L3 𝛿C (ppm) | C3 𝛿C (ppm) |
| Cq =N–OH | 161.51 | 181.64 |
| Cq–(py) | 159.54 | 166.44 |
| CH | 149.35 | 149.80 |
| Cq–CH3 | 148.98 | 146.09 |
| CH(py) | 136.83 | 141.48 |
| CH(py) | 122.34 | 124.99 |
| CH(py) | 122.16 | 123.18 |
| CH2= | 109.69 | 111.89 |
| Cq–NH | 56.54 | 68.33 |
| CH2–NH | 47.94 | 51.46 |
| CH2 | 40.50 | 46.47 |
| CH2–C–NOH | 26.12 | 31.78 |
| CH2 | 25.18 | 29.22 |
| CH3–CNH | 23.84 | 23.79 |
| CH3–C =CH2 | 20.84 | 21.97 |
4.4. X-ray diffraction of C1 and C2 complexes
Single-crystal X-ray measurements were performed at 100 K using an Oxford Cryostream 700 (Oxford Cryosystem). Data for both C1 and C2 complexes were collected on an Apex II CCD 4K Bruker diffractometer equipped with a MoK𝛼 Incoatec microsource (𝜆 = 0.71073 Å). The structures were solved using SHELXT [24] and refined by least-squares procedures on F2 using SHELXL [25]. The most important details concerning crystal data, experiment and refinement are summarized in Supporting Information.
4.5. General procedure for allylic C–H alkylation
The precursor Pd2(C3H5)2Cl2(1 mol%), the ligand Ln(2 mol%), K2CO3 (100 mol%) and the solvent (2 mL) were placed in a N2-purged Schlenk tube. The mixture was stirred at 60 °C for 17 h. Conversions and selectivites were determined by gas chromatography (GC) on Shimadzu 2010 equipped with a column Equity-1 (30 m, i.d. = 0.32 mm). The solvent was then evaporated and the crude product was purified by column chromatography. The separations were conducted on silica gel with petroleum/ethyl acetate (98:2) as eluent, affording the desired product.
Conflict of interest
Authors have no conflict of interest to declare.
Acknowledgements
We acknowledge the Ministère de l’Enseignement supérieur de la Recherche et de l’Innovation (France) and the Ministère de la recherche (Maroc) for their financial support. We also would like to thank Céline Delabre for the chromatography analyses.
The Chevreul Institute (FR 2638), Ministry of Higher Education, Research and Innovation, Région Hauts de France and FEDER are recognized for funding of X-ray diffractometers.