


 CC-BY 4.0
CC-BY 4.0
1. Introduction
In 1979, Suzuki and Miyaura reported for the first time the palladium-catalyzed cross-coupling of alkenyl boranes with alkenyl halides in the presence of a base [1]. Since this discovery, the Suzuki–Miyaura cross-coupling reaction has become the preferred method for preparing biaryls by Csp2–Csp2 bond formation, with extensive applications in the synthesis of a wide range of natural products, pharmaceuticals, polymers, and advanced materials [2, 3, 4, 5, 6, 7, 8, 9, 10]. Initially, tetrakis(triphenylphosphine)palladium(0) was employed as catalyst [11], and since then, much effort has been made to develop more efficient catalytic systems able to accelerate the rate of the cross-coupling and/or substitute brominated derivatives with less reactive compounds such as their chlorinated equivalents or acyl electrophiles [12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26]. Let us note in particular the use of bisphosphines [27, 28], sterically hindered N-heterocyclic carbenes [29, 30, 31, 32, 33] or phosphines [34, 35, 36, 37, 38, 39] and hemilabile biarylphosphines [40, 41, 42, 43] as ligands. Interestingly, several nitrogenated ligands have been also employed for the Suzuki–Miyaura cross-coupling. We can quote for example the use of Schiff bases [44, 45, 46, 47, 48, 49, 50], amines [51, 52], azo compounds [53], pyridines [54, 55, 56, 57, 58], pyrimidines [59, 60], triazoles [61] or benzimidazoles [62]. However, despite their relevance, these catalytic systems suffer from one or more drawbacks such as the use of expensive or difficultly accessible ligands, as well as high catalyst loading, hazardous solvents and harsh reaction conditions. Therefore, the development of new, efficient catalytic systems for the Suzuki–Miyaura cross-coupling is still desirable.
In a recent study, we have described the synthesis of three diethyl[(5-phenyl-1,3,4-oxadiazol-2-ylamino)(aryl)methyl]phosphonates (1–3; Figure 1), in which, according to natural bond orbital charge analyzes, the nitrogen atom in position 3 of the oxadiazole ring is the more basic one [63]. Coordination of these 𝛼-aminophosphonates to a ruthenium precursor occurred via its electron-enricher nitrogen atom as confirmed by X-ray crystallographic structure of the [RuCl2(L)(𝜂6-p-cymene)] complexes (L = 1–3). In continuation of our research, we now wish to investigate the ability of the 1,3,4-oxadiazole moiety to coordinate to a palladium precursor and the ability of the 𝛼-aminophosphonates to act as ligand in the Suzuki–Miyaura cross-coupling of aryl halides. The presence of a phosphonate moiety in these ligands may promote the formation of (N,O)-chelate intermediates, which would increase the steric hindrance and the electronic density of the metal and therefore favor the oxidative addition and the reductive elimination steps of the cross-coupling reaction.
To the best of our knowledge, no mention of 1,3,4-oxadiazole derivatives as ligands, in which a nitrogen atom is directly coordinated to palladium, has been reported for the Suzuki–Miyaura cross-coupling. The functionalization of the 1,3,4-oxadiazole ring by the Suzuki–Miyaura methodology has, however, been described for example by the groups of Khazi [64, 65] and Kudelko [66].
Oxadiazole-containing 𝛼-amino- phosphonates ligands (1–4) used in this study.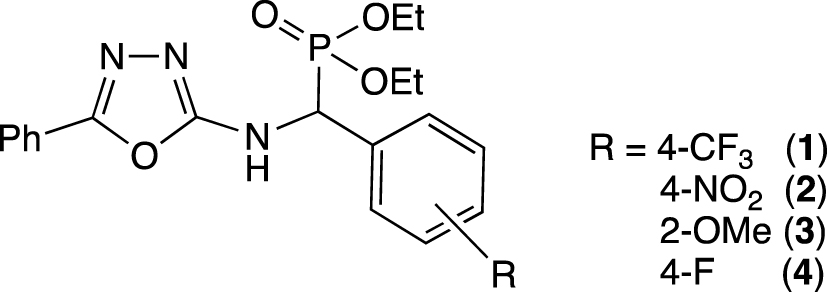
2. Results and discussion
2.1. Synthesis of palladium complex
The new diethyl[(5-phenyl-1,3,4-oxadiazol-2-ylam- ino)(aryl)methyl]phosphonate (4), used in this study, was prepared according to a procedure previously reported by our group [63], which involves a Pudovik-type reaction [67, 68] between diethyl phosphite and imine obtained from 5-phenyl-1,3,4-oxadiazol-2-amine and 4-fluorobenzaldehyde, under microwave irradiation.
Synthesis of trans-[PdCl2(2)2] complex (5).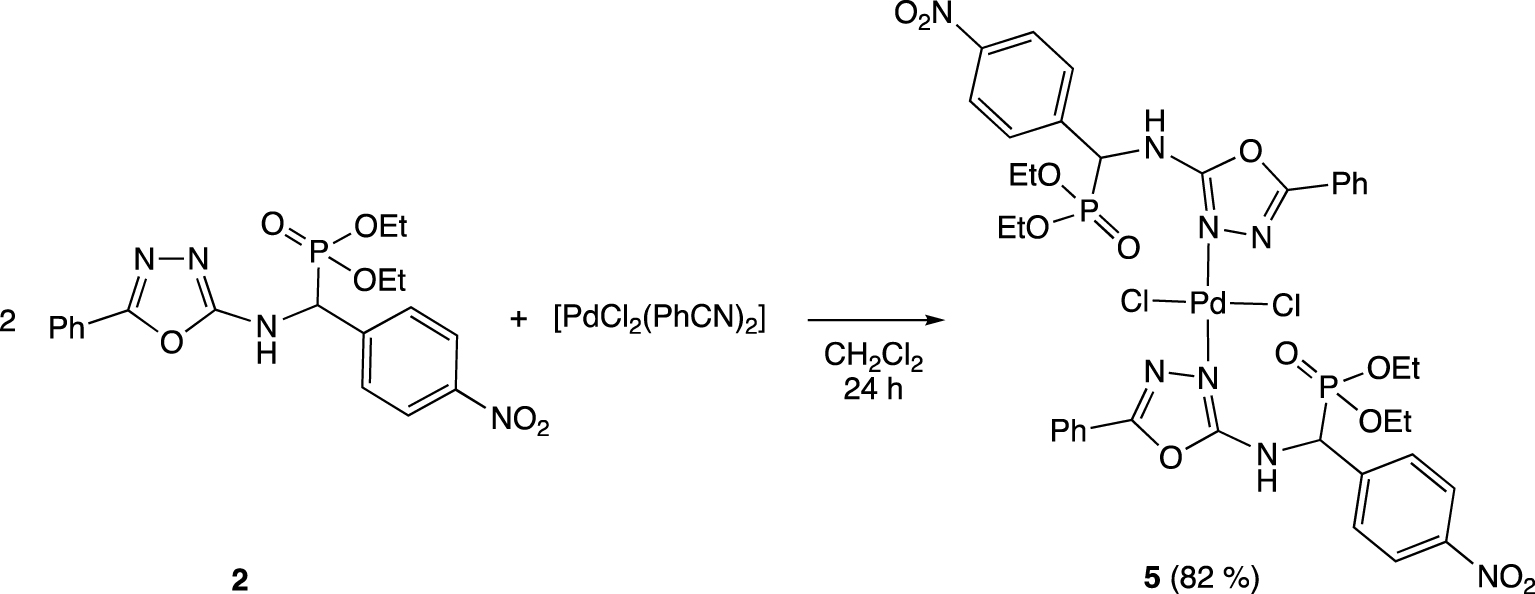
To gain insight into the coordinative properties of these 𝛼-aminophosphonates 1–4, we investigated the coordination of 2 with the [PdCl2(PhCN)2] precursor in dichloromethane for 24 h (Scheme 1). After recrystallization, the [PdCl2(2)2] complex (5) was isolated in 82% yield as a yellow solid. Its 1H NMR spectrum reveals for the CHP(O) proton a downfield shift of 0.1 ppm while the 31P NMR spectrum shows a singlet for the P(O) signal with a slight up field shift of 0.6 ppm with regard to the free 𝛼-aminophosphonate 2. Infrared measurements show that the band at 1601 cm−1 in free ligand 2 (𝜈(C=N) stretching vibrations of the oxadiazole ring) shifts to 1658 cm−1 in the palladium complex 5, which confirms the coordination of the 𝛼-aminophosphonate via its oxadiazole ring. The mass spectrum analysis, which shows a peak corresponding to [M − Cl]+ cation with the expected isotopic profile, indicates the coordination of the two 𝛼-aminophosphonates to the metal. The structure of the complex 5 was confirmed by a single crystal X-ray diffraction study, which unambiguously demonstrated the presence of two 𝛼-aminophosphonates moieties trans-coordinated to the palladium center via their electron-enricher nitrogen atoms of the oxadiazole rings denoted N1 in Figure 2.
ORTEP drawing of complex 5 showing 50% probability thermal ellipsoids. Important bond lengths (Å) and angles (°): Pd1–Cl1 2.3012(5), Pd1–N1 2.0050(16), N1–N2 1.400(2), N2–C2 1.285(2), C2–O1 1.378(2), O1–C1 1.350(2), C1–N1 1.309(2), Cl1–Pd1–N1 90.08(5), N1–Pd1–N1 180.00(6), C2–N2–N1 105.40(15), N2–N1–C1 107.72(15), N1–C1–O1 110.88(16), C1–O1–C2 103.29(14), O1–C2–N2 112.69(16).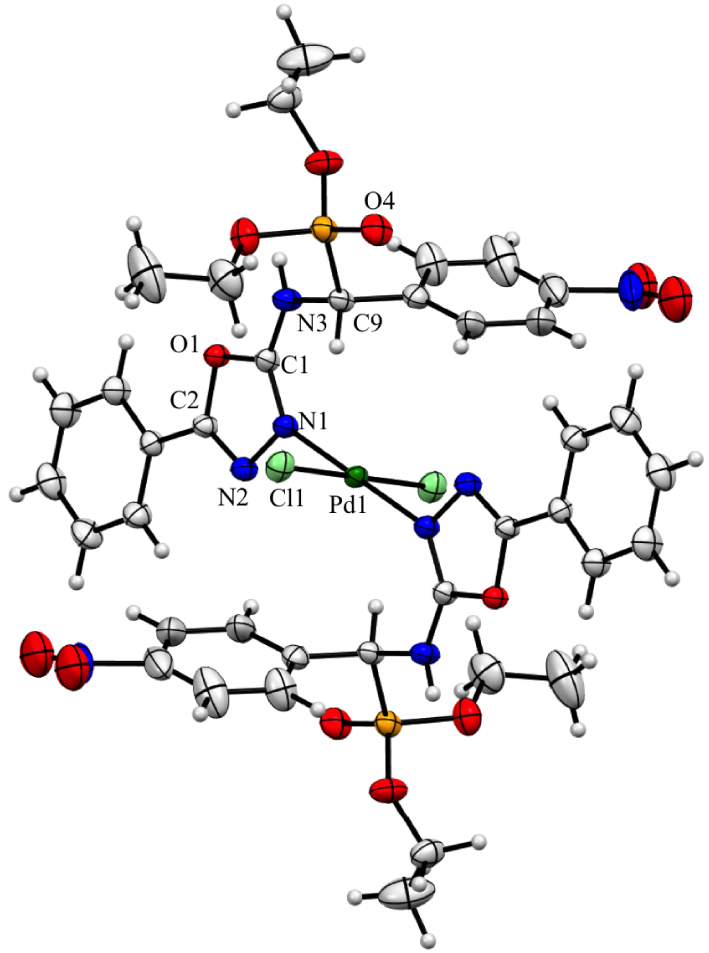
The palladium complex 5 crystallizes in the trigonal space group R-3 with two distinct enantiomeric 𝛼-aminophosphonates (the two C9 atoms have an (R)- and an (S)-configuration). The palladium atom sits on the center of symmetry of the molecule and adopts a square-planar coordination environment with the two 𝛼-aminophosphonates in trans position. The Pd–N and Pd–Cl bond lengths, 2.0050(16) and 2.3012(5) Å, respectively, are similar to those found in the trans-dichloro-bis[2,5-bis(3,4,5-trime- thoxyphenyl)-1,3,4-oxadiazole] palladium(II) [69] or in the dichloro-bis{2-[diazenido-bis(1,2-bis(diphe- nylphosphino)ethane)bromotungsten]-5-(4-cyano- phenyl)-1,3,4-oxadiazole} palladium(II) complexes [70]. The nitro (N4, O2 and O3) and an ethyl (C16 and C17) moieties are disordered over two positions with ratios of 0.6/0.4 and 0.7/0.3, respectively. The oxadiazole and phenyl aromatic rings are nearly planar with a dihedral angles of 4.85° and are almost perpendicular to the aromatic bearing the nitro substituent with dihedral angles of 79.42 and 75.05°, respectively.
2.2. Catalytic Suzuki–Miyaura cross-coupling—the search for optimal catalytic conditions
The four oxadiazole-containing 𝛼-aminophosphonate ligands 1–4 were evaluated in the palladium-catalyzed Suzuki–Miyaura cross-coupling between aryl bromides/chlorides and aryl boronic acids in the presence of a base. For the optimization tests, the catalytic system was generated in situ starting from 𝛼-aminophosphonate 1 and a palladium precursor (0.5 mol%). The tests were carried out using 4-bromoanisole (6a; 0.5 mmol), phenyl boronic acid (7a; 0.75 mmol) and a base (0.75 mmol). The conversions into 4-methoxybiphenyl (8a) and biphenyl, a by-product resulting from the homocoupling of phenyl boronic acid, were determined by gas chromatography after two hours (Scheme 2).
Palladium-catalyzed Suzuki–Miyaura cross-coupling between 4-bromoanisole and phenyl boronic acid.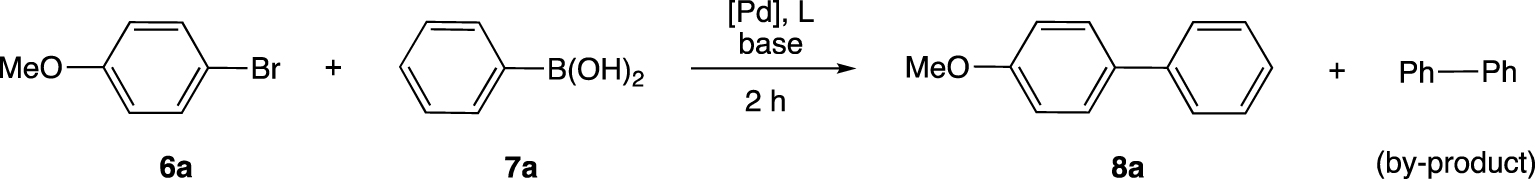
First, we investigated the influence of the palladium source (Table 1). The runs were carried out in 1,4-dioxane as solvent at 100 °C with one equivalent of 𝛼-aminophosphonate 1 per palladium precursor (0.5 mol% each). A screen of four palladium sources, namely [Pd(OAc)2], [PdCl2(PhCN)2], [PdCl2(cod)] and [Pd2(dba)3] (cod = 1,5-cyclooctadiene and dba = dibenzylideneacetone), revealed that [Pd(OAc)2] outperformed other precursors. Despite, the conversion into the expected 4-methoxybiphenyl (8a) is less important than with [PdCl2(cod)], the formation of the by-product remains negligible in the case of [Pd(OAc)2] (Table 1).
Suzuki–Miyaura cross-coupling of 4-bromoanisole with phenyl boronic acid; the search for optimal catalytic conditions: effect of palladium sourcea
| Entry | Palladium precursor | Conversion into 8a (%) | By-product (Ph–Ph) Ph–Ph/8a (%) |
|---|---|---|---|
| 1 | [Pd(OAc)2] | 65 | Traces |
| 2 | [PdCl2(PhCN)2] | 64 | 10 |
| 3 | [PdCl2(cod)] | 74 | 27 |
| 4 | [Pd2(dba)3] | 21 | 11 |
Conditions: apalladium precursor (2.5 × 10−3 mmol, 0.5 mol%), 1 (0.0011 g, 2.5 × 10−3 mmol, 0.5 mol%). MeO-C6H4Br (0.093 g, 0.5 mmol), PhB(OH)2 (0.091 g, 0.75 mmol), Cs2CO3 (0.325 g, 0.75 mmol), 1,4-dioxane (2 mL), decane (0.025 mL), 100 °C, 2 h. The conversions were determined by GC, the calibration being based on decane.
In a second series of catalytic tests, we investigated the influence of the base (Table 2). The runs were carried out in 1,4-dioxane as solvent at 100 °C with one equivalent of 𝛼-aminophosphonate 1 per [Pd(OAc)2] precursor. Four bases derived from carbonate anion were tested, as attempts, and the conversions increased with the size of the cations from lithium (traces; Table 2, entry 1) to cesium (65%; Table 2, entry 4). Interestingly, with the latter base, only traces of by-product were observed. Testing other sodium or potassium bases containing hydroxy, acetate or alkoxide anions did not improve the amount of 4-methoxybiphenyl 8a (Table 2, entries 5–10). Reducing the temperature to 80 °C led to a lower rate of the reaction and a conversion of only 38% was measured (Table 2, entry 11). Changing 1,4-dioxane to THF, DMSO or DMF favored the formation of homocoupling by-product with Ph–Ph/8a ratio up to 88% when DMSO was employed (Table 2, entries 12–14).
Suzuki–Miyaura cross-coupling of 4-bromoanisole with phenyl boronic acid; the search for optimal catalytic conditions: effect of base, solvent and temperaturea
| Entry | Base | Solvent | T (°C) | Conversion into 8a (%) | By-product (Ph–Ph) Ph–Ph/8a (%) |
|---|---|---|---|---|---|
| 1 | Li2CO3 | 1,4-dioxane | 100 | Traces | / |
| 2 | Na2CO3 | 1,4-dioxane | 100 | 18 | Traces |
| 3 | K2CO3 | 1,4-dioxane | 100 | 40 | 20 |
| 4 | Cs2CO3 | 1,4-dioxane | 100 | 65 | Traces |
| 5 | NaOH | 1,4-dioxane | 100 | 26 | 17 |
| 6 | KOH | 1,4-dioxane | 100 | 35 | 8 |
| 7 | NaOAc | 1,4-dioxane | 100 | 20 | Traces |
| 8 | KOAc | 1,4-dioxane | 100 | 24 | / |
| 9 | NaOEt | 1,4-dioxane | 100 | Traces | / |
| 10 | tBuOK | 1,4-dioxane | 100 | 27 | 15 |
| 11 | Cs2CO3 | 1,4-dioxane | 80 | 38 | Traces |
| 12 | Cs2CO3 | THF | 66 | 24 | 10 |
| 13 | Cs2CO3 | DMSO | 100 | 6 | 88 |
| 14 | Cs2CO3 | DMF | 100 | 74 | 16 |
Conditions: a[Pd(OAc)2] (0.0006 g, 2.5 × 10−3 mmol, 0.5 mol%), 1 (0.0011 g, 2.5 × 10−3 mmol, 0.5 mol%), MeO-C6H4Br (0.093 g, 0.5 mmol), PhB(OH)2 (0.091 g, 0.75 mmol), base (0.75 mmol), solvent (2 mL), decane (0.025 mL), 2 h. The conversions were determined by GC, the calibration being based on decane.
Finally, we investigated the influence of the 𝛼-aminophosphonate as well as the metal/ligand ratio (Table 3). The variation of the substituent of the aromatic ring of the 𝛼-aminophosphonate favors the formation of the homocoupling by-product in the order 4-CF3 (1), 4-NO2 (2), 2-MeO (3) and 4-F (4) (Table 3, entries 1–4). With the latter ligand (4), although the conversion of 6a into the cross-coupling product 8a was the more important (76%), the biphenyl by-product was observed in a Ph–Ph/8a ratio of 39% (Table 2, entry 4). Unfortunately, the use of an excess of 𝛼-aminophosphonate 1 per palladium atom failed to produce a selective active species. Although the formation of the desired 4-methoxybiphenyl 8a was favored, the amount of homocoupling by-product was increased (Table 3, entries 5–8). For example, with three ligands per palladium, the conversion reached 85% after 2 hours with a Ph–Ph/8a ratio of 18% (Table 3, entry 8). Interestingly, performing the arylation of 4-bromoanisole with well-defined [PdCl2(2)2] (5) led to conversion similar to that obtained by mixing in situ [Pd(OAc)2] with two equivalents of 𝛼-aminophosphonate 2 (Table 3, entries 9 and 10). Carrying out the experiment using Cs2CO3 in 1,4-dioxane at 100 °C in the presence of a drop of mercury did not significantly change the catalytic outcome, which implies that the cross-coupling reaction does not involve palladium nanoparticles as heterogeneous active species (Table 3, entry 11) [38]. Finally, the control experiment performed in free 𝛼-aminophosphonate condition led to the formation of the desired product 8a in only 15% conversion (Table 3, entry 12). As attempt, no conversion was detected when the runs was repeated in the absence palladium source (Table 3, entry 13). These results suggested that the active species should be a palladium complex coordinated to only one 𝛼-aminophosphonate. It is well established that the presence of a hemilabile group, here the P(O)(OEt)2 moiety, could temporarily bind the metal, making the metal environment sterically more crowded and more electron-rich [71, 72, 73, 74, 75]. These two elements promote the oxidative addition and the reductive elimination/product decoordination elementary steps [76].
2.3. Catalytic Suzuki–Miyaura cross-coupling—comparison with other catalytic systems
To rank the 𝛼-aminophosphonate 1, runs were carried out with benchmark ligands. These experiments were performed with 4-bromoanisole and phenyl boronic acid in the optimized catalytic conditions. For these comparative tests, triphenylphosphine (PPh3), (o-anisyl)diphenylphosphine (L1), 2-diphenylphosphano-2′-methylbiphenyl (L2), 1,2-bis(diphenylphosphino)ethane (dppe), 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (L3) and the calixarenyl-PEPPSI-type complex (Pd1) [77] were employed (Figure 3).
Ligands and palladium complex used to rank the 𝛼-aminophosphonate 1.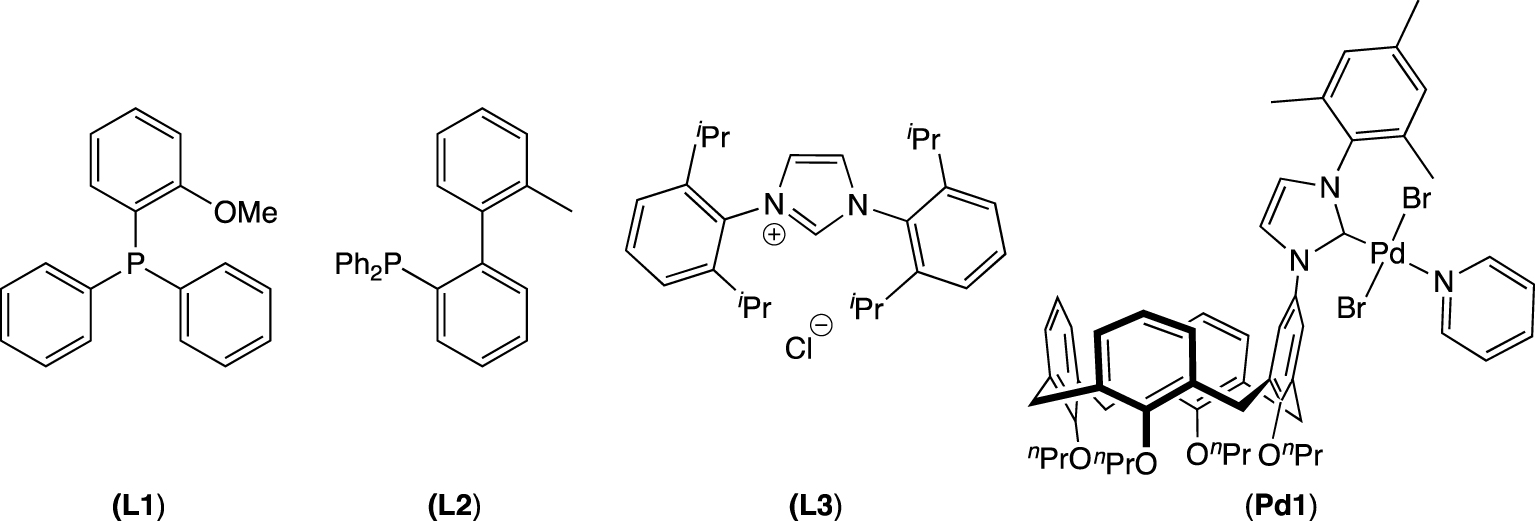
Suzuki–Miyaura cross-coupling of 4-bromoanisole with phenyl boronic acid; the search for optimal catalytic conditions: effect of ligand and metal/ligand ratioa
| Entry | Palladium precursor | Ligand (equiv. /Pd) | Conversion 8a (%) | By-product (Ph–Ph) Ph–Ph/8a (%) |
|---|---|---|---|---|
| 1 | [Pd(OAc)2] | 1 (1) | 65 | Traces |
| 2 | [Pd(OAc)2] | 2 (1) | 44 | 10 |
| 3 | [Pd(OAc)2] | 3 (1) | 69 | 16 |
| 4 | [Pd(OAc)2] | 4 (1) | 76 | 39 |
| 5 | [Pd(OAc)2] | 1 (1.5) | 69 | 7 |
| 6 | [Pd(OAc)2] | 1 (2) | 73 | 16 |
| 7 | [Pd(OAc)2] | 1 (2.5) | 80 | 16 |
| 8 | [Pd(OAc)2] | 1 (3) | 85 | 18 |
| 9b | [PdCl2(2)2] (5) | / | 56 | 13 |
| 10 | [Pd(OAc)2] | 2 (2) | 53 | 14 |
| 11c | [Pd(OAc)2] | 1 (1) | 63 | 4 |
| 12d | [Pd(OAc)2] | / | 15 | 12 |
| 13e | / | 1 (1) | 0 | 0 |
Conditions: apalladium precursor (2.5 × 10−3 mmol, 0.5 mol%), ligand, MeO-C6H4Br (0.093 g, 0.5 mmol), PhB(OH)2 (0.091 g, 0.75 mmol), Cs2CO3 (0.325 g, 0.75 mmol), 1,4-dioxane (2 mL), decane (0.025 mL), 100 °C, 2 h. The conversions were determined by GC, the calibration being based on decane; bwithout additional ligand 2; cwith a drop of mercury; dwithout ligand; ewithout palladium precursor.
As can be seen in Table 4, the performance of 𝛼-aminophosphonate 1 surpasses that of PPh3 or imidazolium salt (L3) and is similar to that of dppe (Table 4, entries 1, 2, 5 and 6). However, the conversion is lower than those measured when (o-anisyl)diphenylphosphine L1 and Buchwald-type biarylphosphine L2 associated with [Pd(OAc)2] and pyridine-enhanced precatalyst Pd1 were employed (Table 4, entries 3, 4 and 7).
2.4. Catalytic Suzuki–Miyaura cross-coupling of aryl halides
Having optimized conditions in hand, [Pd(OAc)2], ligand 1 (Pd/1 : 1/1), Cs2CO3, 1,4-dioxane at 100 °C, seven aryl bromides, namely 4-bromoanisole (6a), 2-bromoanisole (6b), 2-bromo-6-methoxynaphthalene (6c), 4-bromotoluene (6d), 3-bromotoluene (6e), 2-bromotoluene (6f) and 1-bromonaphthalene (6g), were assigned into the cross-coupling reaction with three aryl boronic acids 7a–c for 5 hours (Scheme 3 and Figure 4).
Palladium-catalyzed Suzuki–Miyaura between various aryl halides 6a–k and aryl boronic acids 7a–e.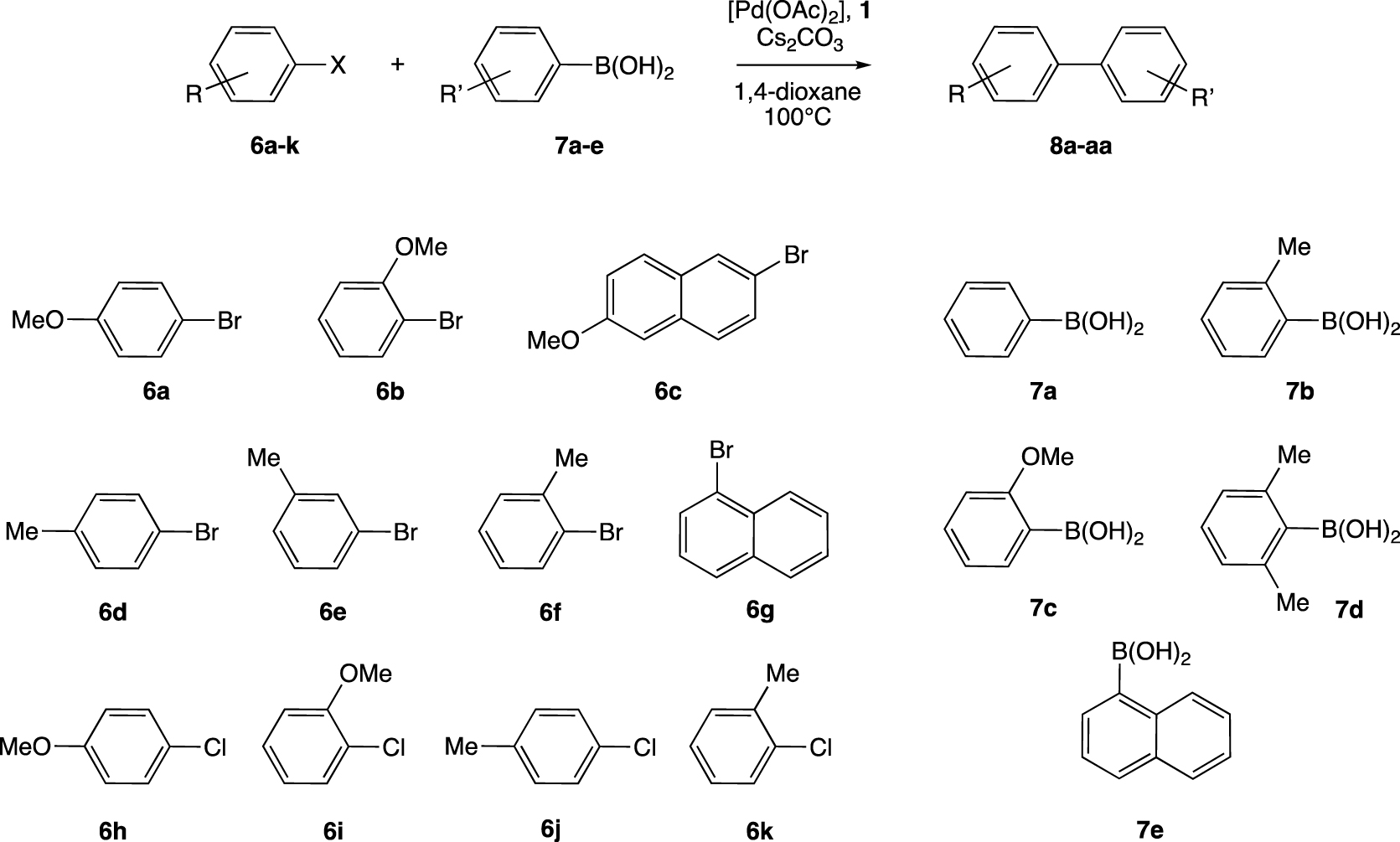
Suzuki–Miyaura cross-coupling of 4-bromoanisole with phenyl boronic acid; the comparison with classical catalytic systemsa
| Entry | Ligand | Conversion 8a (%) | By-product (Ph–Ph) Ph–Ph/8a (%) |
|---|---|---|---|
| 1 | 1 | 65 | Traces |
| 2 | PPh3 | 55 | 6 |
| 3 | L1 | 72 | 2 |
| 4 | L2 | 97 | 2 |
| 5 | dppe | 67 | 4 |
| 6 | L3 | 49 | Traces |
| 7b | / | 86 | 1 |
Conditions: a[Pd(OAc)2] (2.5 × 10−3 mmol, 0.5 mol%), ligand (2.5 × 10−3 mmol, 0.5 mol%), MeO-C6H4Br (0.093 g, 0.5 mmol), PhB(OH)2 (0.091 g, 0.75 mmol), Cs2CO3 (0.325 g, 0.75 mmol), 1,4-dioxane (2 mL), decane (0.025 mL), 100 °C, 2 h. The conversions were determined by GC, the calibration being based on decane; busing calixarenyl-PEPPSI-type complex Pd1 (2.5 × 10−3 mmol, 0.5 mol%) without additional ligand.
Palladium-catalyzed Suzuki–Miyaura cross-coupling: formation of biphenyls from aryl bromides and aryl boronic acids; reagents and conditions: [Pd(OAc)2] (0.5 mol%), 1 (0.5 mol%), ArBr (0.5 mmol), Ar′B(OH)2 (0.75 mmol), Cs2CO3 (0.75 mmol), 1,4-dioxane (2 mL), decane (0.025 mL), 5 h, 100 °C. The conversions were determined by GC, the calibration being based on decane.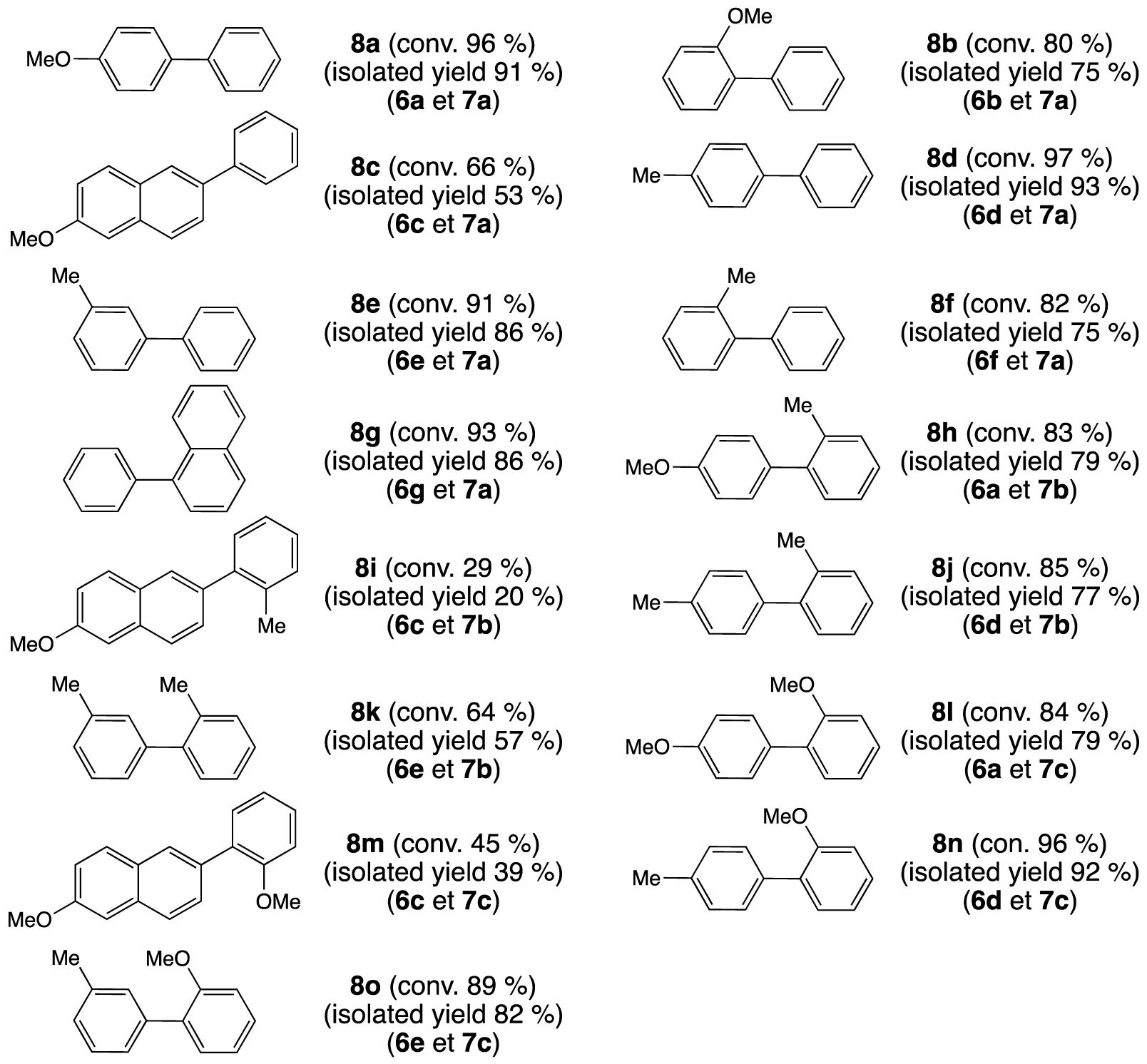
Cross-coupling using the latter compounds led, generally, to high conversions, up to 97% in the case of 4-methylbiphenyl (8d), except when 2-bromo-6-methoxynaphthalene (6c) was employed, which led to modest conversions of 29 and 45% with sterically hindered aryl boronic acids 7b and 7c, respectively. Important reactivities were observed for the formation of ortho-substituted biphenyls, for example, 1-phenylnaphthalene (8g) and 2-methoxy-4′-methylbiphenyl (8n) are formed with conversions of 93 and 96%, respectively. We further found that the homocoupling by-product, coming from aryl boronic acid 7a–c (Ar′B(OH)2), was also formed. However, the Ar′–Ar′/Ar–Ar′ ratio never exceeded 5%.
A longer reaction time, 16 hours at 100 °C, allowed us to obtain ortho-di-/trisubstituted biphenyls 8p–y with conversions in the ranges of 30–84 and 46–68%, respectively (Figure 5). As expected, 6c led to the lowest formation of the coupling product 8s. Surprisingly, the extremely hindered boronic acid 7d generated the ortho-di/trisubstituted biphenyls derivatives with good conversions up to 84% for the ortho-disubstituted biphenyl 8q and 68% for the ortho-trisubstituted biphenyl 8r.
Palladium-catalyzed Suzuki–Miyaura cross-coupling: formation of ortho-di/trisubstituted biphenyls from aryl bromides and aryl boronic acids; reagents and conditions: [Pd(OAc)2] (0.5 mol%), 1 (0.5 mol%), ArBr (0.5 mmol), Ar′B(OH)2 (0.75 mmol), Cs2CO3 (0.75 mmol), 1,4-dioxane (2 mL), decane (0.025 mL), 16 h, 100 °C. The conversions were determined by GC, the calibration being based on decane.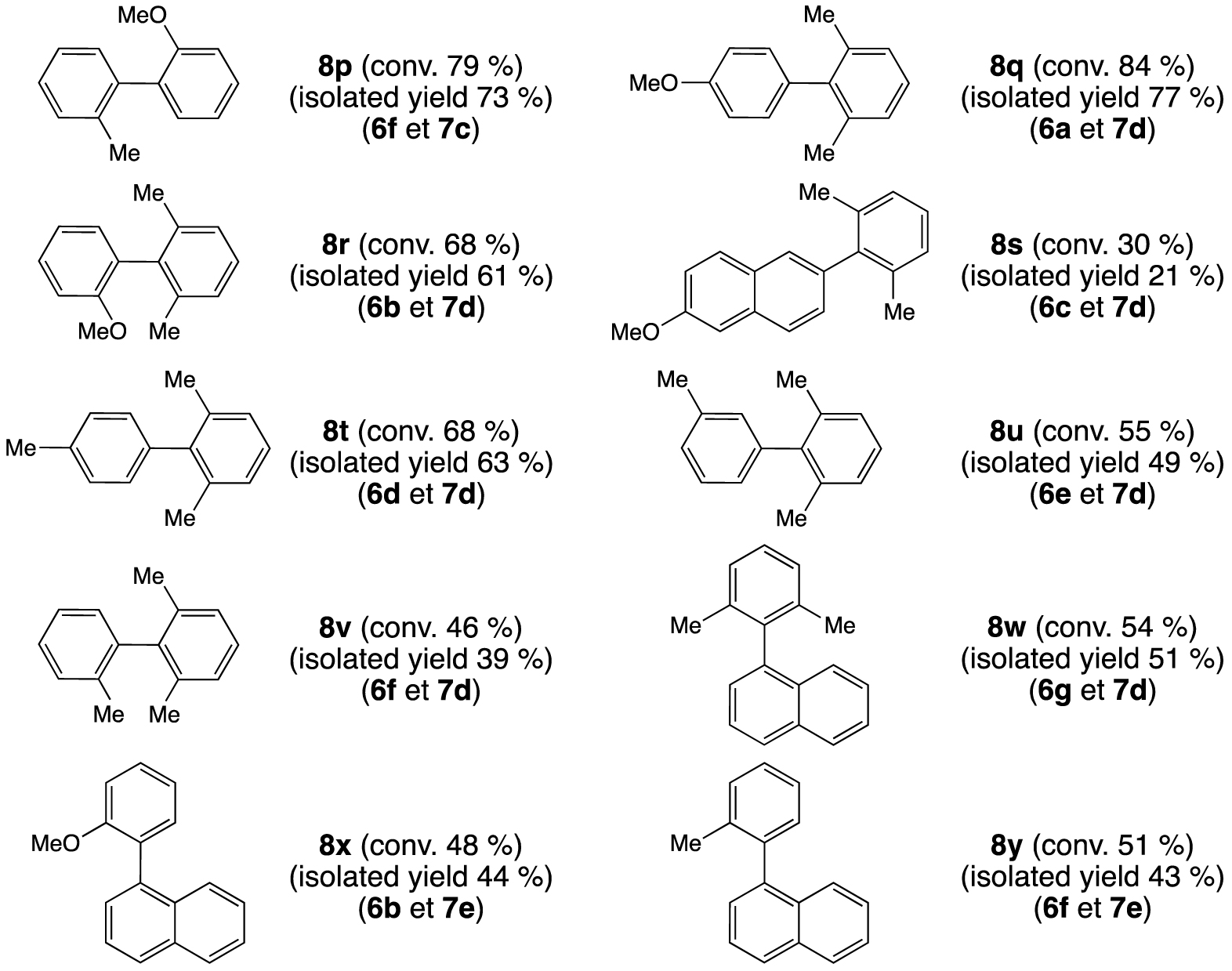
The more challenging aryl chlorides, namely 4-chloroanisole (6h), 2-chloroanisole (6i), 4-chloro- toluene (6j), and 2-chlorotoluene (6k) (Scheme 3), can efficiently undergo Suzuki–Miyaura cross-coupling when these reactions were achieved with a larger catalyst loading, 1 mol% of [Pd(OAc)2] (Figure 6). In these conditions, high amounts of 4-methoxy and 4-methylbiphenyl (8a and 8c) were obtained with conversions of 65 and 78%, respectively. The catalytic system even enabled the formation of ortho-substituted biphenyl with conversions up to 62% when 4-chlorotoluene (6j) and 2-methylphenyl boronic acid (7b) were employed.
Palladium-catalyzed Suzuki–Miyaura cross-coupling: formation of biphenyls from aryl chlorides and aryl boronic acids; reagents and conditions: [Pd(OAc)2] (1 mol%), 1 (1 mol%), ArCl (0.5 mmol), Ar′B(OH)2 (0.75 mmol), Cs2CO3 (0.75 mmol), 1,4-dioxane (2 mL), decane (0.025 mL), 16 h, 100 °C. The conversions were determined by GC, the calibration being based on decane.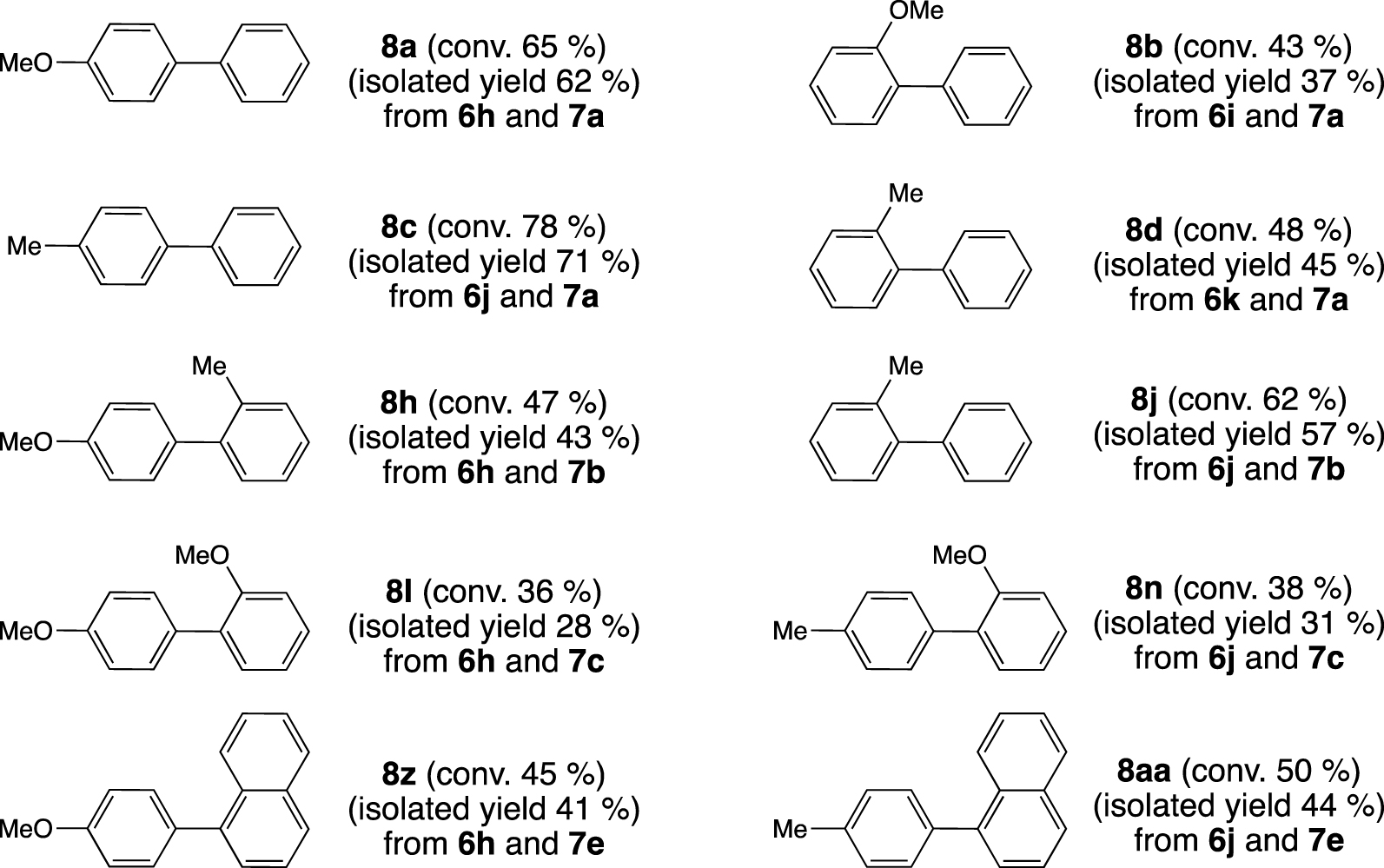
3. Conclusion
We have shown in this work that 𝛼-aminophos- phonates possessing 1,3,4-oxadiazole moieties are able to coordinate to a palladium(II) ion via their electronically enriched nitrogen atom. For the first time, these ligands associated with a palladium precursor, have been shown to be efficient catalytic systems for the Suzuki–Miyaura cross-coupling between aryl bromides or chlorides and aryl boronic acids. A metal loading of 0.5 mol% could be sufficient for the formation of ortho-di/trisubstituted biphenyls with good conversions. The performances of these catalytic systems are probably due to the presence in the 𝛼-aminophosphonates of a hemilabile P(O)(OEt)2 moiety, able to generate (N,O)-chelate palladium complexes. The formation of such species should increase the steric hindrance and the electronic density of the metal, which promote both the oxidative addition and the reductive elimination/product decoordination elementary steps and in fine accelerate the cross-coupling reaction. Further study is aimed at investigating and exploiting in homogeneous catalysis the hemilability of these specific oxadiazole-containing 𝛼-aminophosphonates ligands.
4. Experimental section
All manipulations involving 𝛼-aminophosphonates were carried out under dry argon. Solvents were dried by conventional methods and were distilled immediately before use. Routine 1H, 13C{1H}, 31P{1H} and 19F{1H} spectra were recorded with Bruker FT instruments (AC300 and 500). 1H NMR spectra were referenced to residual protonated solvents (𝛿 = 2.50 ppm for DMSO-d6 and 𝛿 = 7.26 ppm for CDCl3). 13C NMR chemical shifts are reported relative to deuterated solvents (𝛿 = 39.52 ppm for DMSO-d6 and 𝛿 = 77.16 ppm for CDCl3). 31P and 19F NMR spectroscopic data are given relative to external H3PO4 and CCl3F, respectively. Chemical shifts and coupling constants are reported in ppm and Hz, respectively. Infrared spectra were recorded with a Bruker FT-IR Alpha-P spectrometer. Microwave irradiation was carried out using the CEM Discover microwave synthesis system equipped with a pressure controller (17 bar). Elemental analyzes were carried out by the Service de Microanalyse, Institut de Chimie, Université de Strasbourg. 5-Phenyl-1,3,4-oxadiazol-2-amine [78], diethyl[(5- phenyl-1,3,4-oxadiazol-2-ylamino)(4-trifluorometh- ylphenyl)methyl] phosphonate [63] (1), diethyl[(5- phenyl-1,3,4-oxadiazol-2-ylamino)(4-nitrophenyl) methyl]phosphonate [63] (2) and diethyl[(5-phenyl-1,3,4-oxadiazol-2-ylamino)(2-methoxyphenyl)met- hyl]phosphonate [63] (3) were prepared by literature procedures.
4.1. Synthesis of diethyl[(5-phenyl-1,3,4- oxadiazol-2-ylamino)(aryl)methyl] phosphonate (4)
The synthesis of 4 required, in a first step the preparation of (E)-1-(4-fluorophenyl)-N-(5-phenyl-1,3,4-oxadiazol-2-yl)methanimine. In a round bottomed flask equipped with a Dean Stark apparatus, a mixture of phenyl-1,3,4-oxadiazol-2-amine (2.0 mmol) and 4-fuorobenzaldehyde (3.0 mmol) in toluene (20 mL) was refluxed for 48 h. After cooling to room temperature, the solvent was evaporated and the crude product was washed with cool ethanol (15 mL), filtered and dried under vacuum to afford the desired imine in yield 75% (0.401 g) as a white solid. 1H NMR (300 MHz, DMSO-d6): 𝛿 = 9.36 (s, 1H, N=CH), 8.22 (d, 1H, arom. CH, 3J = 8.7 Hz), 8.20 (d, 1H, arom. CH, 3J = 8.7 Hz), 8.08–8.05 (m, 2H, arom. CH), 7.66–7.60 (m, 3H, arom. CH), 7.49 (d, 1H, arom. CH, 3J = 8.7 Hz), 7.46 (d, 1H, arom. CH, 3J = 8.7 Hz) ppm; 13C{1H} NMR (126 MHz, DMSO-d6): 𝛿 = 168.91 (s, N=CH), 166.11 (s, arom. Cquat C(N)=N), 165.64 (d, arom. Cquat CF, 1JCF = 254.4 Hz), 162.83 (s, arom. Cquat C(Ph)=N), 133.09 (d, arom. CH, 3JCF = 9.7 Hz), 132.08 (s, arom. CH of C6H5), 131.10 (d, arom. Cquat of C6H4F, 4JCF = 3.9 Hz), 129.48 (s, arom. CH of C6H5), 126.42 (s, arom. CH of C6H5), 123.54 (s, arom. Cquat of C6H5), 116.67 (d, arom. CH, 2JCF = 22.3 Hz) ppm; 19F{1H} NMR (282 MHz, DMSO-d6): 𝛿 = −103.79 (s, CF) ppm. Elemental analysis calcd. (%) for C15H10ON3F (267.26): C 67.41, H 3.77, N 15.72; found C 67.46, H 3.74 N 15.67.
In a second step, in a round bottomed flask, a mixture of imine (1.0 mmol) and diethyl phosphite (2.0 mmol) was added and irradiated under microwave in neat condition at 115 °C for 10 min at 300 W. After cooling to room temperature, the crude product was washed with Et2O (3 × 10 mL), filtered and dried under vacuum to afford the desired 𝛼-aminophosphonate in yield 97% (0.393 g) as a white solid. 1H NMR (300 MHz, DMSO-d6): 𝛿 = 9.00 (dd, 1H, NH, 3JHH = 10.2 Hz, 3JPH = 3.3 Hz), 7.85–7.81 (m, 2H, arom. CH), 7.64–7.58 (m, 2H, arom. CH), 7.56–7.51 (m, 3H, arom. CH), 7.24 (d, 1H, arom. CH, 3JHH = 8.7 Hz), 7.21 (d, 1H, arom. CH, 3JHH = 8.8 Hz), 5.24 (dd, 1H, CHP, 2JPH = 21.6 Hz, 3JHH = 9.9 Hz), 4.09–3.81 (m, 4H, OCH2CH3), 1.17 (t, 3H, OCH2CH3, 3JHH = 7.0 Hz), 1.08 (t, 3H, OCH2CH3, 3JHH = 7.0 Hz) ppm; 13C{1H} NMR (126 MHz, DMSO-d6): 𝛿 = 163.01 (d, arom. Cquat of C6H4F, 2JCP = 11.0 Hz), 161.83 (d, arom. Cquat CF, 1JCF = 243.8 Hz), 158.30 (s, arom. Cquat C(Ph)=N), 131.68 (s, arom. Cquat C(NH)=N), 130.78 (s, arom. CH of C6H5), 130.33 (d, arom. CH of C6H4F, 3JCF = 5.9 Hz), 130.28 (d, arom. CH of C6H4F, 3JCF = 7.6 Hz), 129.32 (s, arom. CH of C6H5), 125.29 (s, arom. CH of C6H5), 123.98 (s, arom. Cquat of C6H5), 115.12 (d, arom. CH of C6H4F, 2JCF = 21.4 Hz), 62.54 (d, CH2CH3, 2JCP = 13.7 Hz), 62.69 (d, CH2CH3, 2JCP = 13.5 Hz), 53.66 (d, CHP, 1JCP = 155.5 Hz), 16.26 (d, CH2CH3, 3JCP = 5.0 Hz), 16.11 (d, CH2CH3, 3JCP = 5.2 Hz) ppm; 31P{1H} NMR (121 MHz, DMSO-d6): 𝛿 = 20.1 (d, P(O), 5JPF = 4.3 Hz) ppm; 19F{1H} NMR (282 MHz, DMSO-d6): 𝛿 = −114.37 (d, 5JPF = 4.4 Hz) ppm. Elemental analysis calcd. (%) for C19H21O4N3FP (405.36): C 56.30, H 5.22, N 10.37; found C 56.16, H 5.19, N 10.31.
4.2. Synthesis of dichloro-bis-{diethyl[(5-phenyl- 1,3,4-oxadiazol-2-ylamino)(4-nitrophenyl) methyl]phosphonate} palladium(II) (5)
In a Schlenk tube under argon atmosphere, a dichloromethane solution (5 mL) made of diethyl[(5-phenyl-1,3,4-oxadiazol-2-ylamino)(4-nitrophenyl)methyl]phosphonate (2) (0.104 g, 0.24 mmol) and [PdCl2(PhCN)2] (0.046 g, 0.12 mmol) was stirred at room temperature for 24 h. The reaction mixture was then evaporated under reduced pressure to dryness. The desired compound was purified by slow diffusion of hexane into a dichloromethane solution of the crude product. The solid was filtered, washed with hexane and dried under vacuum to afford complex 5 as a yellow solid in 82% yield (0.102 g). 1H NMR (500 MHz, CDCl3): 𝛿 = 8.22 (d, 2H, arom. CH, 3JHH = 8.5 Hz), 7.86 (d, 2H, arom. CH of O2NC6H4, 3JHH = 8.0 Hz), 7.76 (d, 2H, arom. CH of O2NC6H4, 3JHH = 8.0 Hz), 7.49–7.42 (m, 3H, arom. CH), 5.34 (d, 1H, CHP, 2JPH = 22.5 Hz), 4.27–4.16 (m, 2H, OCH2CH3), 4.10–4.02 (m, 1H, OCH2CH3), 3.95–3.86 (m, 1H, OCH2CH3), 1.33 (t, 3H, OCH2CH3, 3JHH = 7.0 Hz), 1.19 (t, 3H, OCH2CH3, 3JHH = 7.0 Hz) ppm; 13C{1H} NMR (126 MHz, CDCl3): 𝛿 = 162.30 (d, arom. Cquat para of NO2, 2JCP = 13.1 Hz), 160.06 (s, arom. Cquat C(Ph)=N), 148.01 (d, arom. Cquat C(NH)=N, 3JCP = 2.6 Hz), 142.33 (s, arom. Cquat CNO2), 131.08 (s, arom. CH), 129.09 (s, arom. CH), 129.05 (s, arom. CH of O2NC6H4), 126.032 (s, arom. CH of O2NC6H4), 124.16 (s, arom. Cquat of C6H5), 123.93 (s, arom. CH), 64.18 (d, OCH2CH3, 2JCP = 6.8 Hz), 64.17 (d, OCH2CH3, 2JCP = 6.9 Hz), 54.96 (d, CHP, 1JCP = 153.5 Hz), 16.58 (d, OCH2CH3, 3JCP = 5.5 Hz), 16.39 (d, OCH2CH3, 3JCP = 5.4 Hz) ppm; 31P{1H} NMR (121 MHz, CDCl3): 𝛿 = 16.7 (s, P(O)) ppm. IR: 𝜈 = 1658 cm−1 (C=N). MS (ESI-TOF): m∕z = 1005.11 [M–Cl]+ (expected isotopic profile). Elemental analysis calcd. (%) for C38H42O12N8P2Cl2Pd (1042.06): C 43.80, H 4.06, N 18.42; found C 43.61, H 4.04, N 18.29.
4.3. General procedure for the palladium-catalyzed Suzuki–Miyaura cross-coupling reactions
In a Schlenk tube, in an inert atmosphere, a solution of [Pd(OAc)2] in 1,4-dioxane, a solution of the ligand 1 (ratio Pd/1 = 1/1) in 1,4-dioxane, aryl halide (0.5 mmol), aryl boronic acid (0.75 mmol), Cs2CO3 (0.325 g, 0.75 mmol), decane (0.025 mL, internal reference) and a complementary amount of 1,4-dioxane, so that the total reaction volume was 2.0 mL, were introduced. The reaction mixture was then heated at 100 °C. After cooling to room temperature, a small amount (0.5 mL) of the resulting solution was passed through a Millipore filter and analyzed by GC.
4.4. X-Ray crystallographic study structure
Single crystals of 5 suitable for X-ray analysis were obtained by slow diffusion of hexane into a dichloromethane solution of the complex. Crystal data: C38H42Cl2N8O12P2Pd, Mr = 1042.06 g⋅mol−1, trigonal, space group R-3, a = 31.9457(8) Å, b = 31.9457(8) Å, c = 11.6517(5) Å, V = 10297.8(7) Å3, Z = 9, D = 1.512 g⋅cm−3, 𝜇 = 0.660 mm−1, F(000) = 4788, T = 120(2) K. The sample (0.300 × 0.280 × 0.250) was studied on a Bruker PHOTON-III CPAD using Mo-K𝛼 radiation (𝜆 = 0.71073 Å). The data collection (2𝜃max = 30.044°, omega scan frames by using 0.7° omega rotation and 30 s per frame, range hkl: h − 44,44 k − 44,44 l − 16,16) gave 80,005 reflections. The structure was solved with SHELXT-2014/5 [79], which revealed the non-hydrogen atoms of the molecule. After anisotropic refinement, all of the hydrogen atoms were found with a Fourier difference map. The structure was refined with SHELXL-2018/3 [80] by the full-matrix least-square techniques (use of F square magnitude; x, y, z, 𝛽ij for C, N, O, P, Cl and PD atoms; x, y, z in riding mode for H atoms); 302 variables and 6445 observations with I > 2.0 𝜎(I); calcd. w = 1∕[𝜎2(Fo2) + (0.0474P)2 + 37.0114P] where P = (Fo2 + 2Fc2)∕3, with the resulting R = 0.0373, RW = 0.1047 and SW = 1.111, 𝛥𝜌 < 1.137 eÅ−3.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgments
We gratefully acknowledge the Tunisian Ministry of Higher Education and Scientific Research for the financial support (grant for SH).