

1 Introduction
The incorporation of C60 in a polymer matrix is an important issue as it allows combining in a single material the very unique electronic properties of this fullerene with the well known advantages of polymers: easy processing, low cost etc. But, the low solubility or incompatibility of C60 in polymers is one of the main drawbacks for many applications where such materials would be of prime importance. Grafting polymer chains onto C60 allows overcoming this limitation. Nevertheless, any addition reaction on one of the 30 6–6 bonds on this fullerene results in the formation of a single bond and thus induces a change in its electronic properties. To produce materials where the properties of the incorporated C60 are well defined, it is essential that the number of grafted chains (i.e. the number of opened double bonds) is perfectly controlled and is identical for all the fullerenes. In addition, as the distance between the C60 cores of stars with a given number of arms will directly depend on the length of the grafted chains, it is also essential to be able to control the molar mass of the polymer and keep its polydispersity as low as possible.
From all the polymerization methods, the anionic [1] and the ‘controlled’ radical polymerizations [2] are the ones that give the best control over the chain length and produce polymers with the lowest polydispersities. In addition, polymers prepared through these routes bear a terminal function that is able to add to one of the ‘double’ bonds (6–6 bonds) of C60: respectively, a carbanion or a bond that can be converted to a radical. Furthermore, it is possible to take advantage of the specific shape and chemical reactivity of C60 to perfectly control the number of polymer chains grafted onto the fullerene using these two types of addition.
In this short review we will discuss the mechanism of addition of ‘living’ anionic polymers as well as that of macro-radicals onto the C60 and we will present a few examples of the ‘model’ architectures that can be prepared. We will also give some information concerning the thermal stability of these materials as that is of prime importance for any application. Finally, we will show how one can take advantage of the presence of grafted polymer chains on the C60 to organize the fullerene at the nanoscale by using the self-organization of block copolymers.
2 Results and discussion
2.1 Grafting of ‘living’ carbanionic polymers onto C60
2.1.1 In non-polar solvents
The addition of t-butyllithium onto C60 was the first addition of carbanions reported [3,4]. If the organolithium is used in excess, a mixture of adducts of various functionalities is obtained but no adduct higher than the hexa-adduct was observed. The addition of polymer chains bearing at one end a carbanion was first reported by Samulski et al. [5]. By reacting polystyryllithium with C60 in toluene (Scheme 1) a mixture of stars with a fullerene core (PS)xC60 was obtained and the functionality x was supposed to range from 1 to 10. This addition reaction of PSLi onto C60 in toluene was also studied by Zgonnik et al. [6] and they concluded that the product obtained was of a mixture of tetra-, hexa- and octa-aducts. Even by using a large excess of PSLi in cyclohexane, Wang et al. [7] report that they could not graft more than two chains and proposed that it was due to steric hindrance. During the same period, we were also interested in grafting PSLi onto C60. Our first important observation was that by working under very clean conditions (in glass apparatus sealed under high vacuum, using the break-seal technique [1]) and by using carefully purified fullerene it was possible to avoid the formation of mixtures and get pure tri-, tetra-, penta- and hexa-adducts just by controlling the stoechiometry PSLi/C60 [8,9]. Pure mono- or di-adducts could not be obtained through this route as the decrease of reactivity of C60 after the first and the second addition is not sufficiently pronounced. Using multiple detector size exclusion chromatography (SEC) we have proven that the highest adduct that can be produced by this route is the hexa-adduct. The quantitative response of the refractive index (RI) detector allowed us to show that, if the excess of PSLi becomes higher than 6, the molar mass of the stars stays constant and the amount of ungrafted chains is exactly what is predicted from the stoechiometry assuming the formation of a pure hexa-adduct [9]. In addition the polydispersity of the stars (Mw/Mn ≤ 1.1) is as low or even lower than that of the starting PS, indicating that there is no mixture of functionality. An example of the SEC analysis of such a hexa-adduct is given in Fig. 1. The response of a UV-detector set at 320 nm, where the PS is not longer detected but where the fullerene containing molecules absorb light, shows that C60 is only present in the peak of highest molar mass attributed to the stars (Fig. 1(3)). Finally, the light scattering (LS) detector allows determining the absolute molar mass of the stars. The results given in Table 1 and in Refs. [8–10] demonstrate that the molar mass of the stars is six times that of the PS arm whatever the molar mass of this latter thus excluding any steric limitation on the functionality. The dimensions of these hexa-adducts in solution are in good agreement with those expected for stars with six arms and, upon decreasing the molar mass of the arm, an evolution from a random coil-type behavior toward that of a dense sphere can be observed [10]. The structural behavior of these six-arm stars with a C60 core has been extensively studied in dilute and semi-dilute solution by small angle neutron scattering and the results will be published soon [11]. The upper limit of six grafted chains when C60 is reacted with a ‘living’ PSLi is a direct consequence of the specific shape of the C60 molecule and the fact that each addition introduces on the fullerene core a negative charge that delocalizes on the conjugated cage. As illustrated in Scheme 2a, the carbanion on the fullerene is delocalized over the pyracyclene unit including in its center the double bond to which the first PSLi adds, so that no further addition can take place on this part of the C60 molecule. Even, the addition of a second ‘living’ chain to one of the double bonds of the four hexagons h1, h2, h3, and h4 surrounding the pyracyclene unit is not likely as it would lead to strong Coulombic repulsive interactions between the two negative charges. Considering the geometry of the C60, the most favorable site for the second addition would be on the central double bond of one of the adjacent pyracyclenes (arrows in Scheme 2b). Indeed, that allows the two carbanions introduced on the C60 to delocalize without direct interference as illustrated in Scheme 2b. Once, a PSLi chain has attached to the central bond of the six pyracyclene units constituting the C60, the whole conjugated fullerene molecule is covered by the six delocalized carbanions (Scheme 2c) and no further addition can take place. This specific addition mechanism allows to prepare PS stars with exactly six arms and a C60 core just by using a PSLi/C60 ration in excess of 6. After quenching of the carbanions on the fullerene core by protonation, the ungrafted chains can be easily removed by classical polymer fractionation. The six-arm stars obtained through this route count among the best-defined polymer stars architectures reported in the literature. For example, it is possible to vary the molar mass of these stars over about three order of magnitudes without changing their functionality. That allowed us to study the effect of the molar mass on the formation of membranes with a honeycomb pore organization upon evaporation of carbon disulfide solutions of (PS)6C60 in a humid atmosphere (Fig. 2) [12].
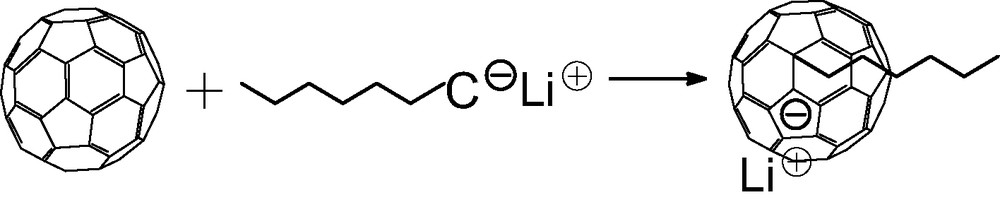
Addition of ‘living’ anionic polymers such as polystyrillithium or PILi onto C60 in a non-polar solvent.
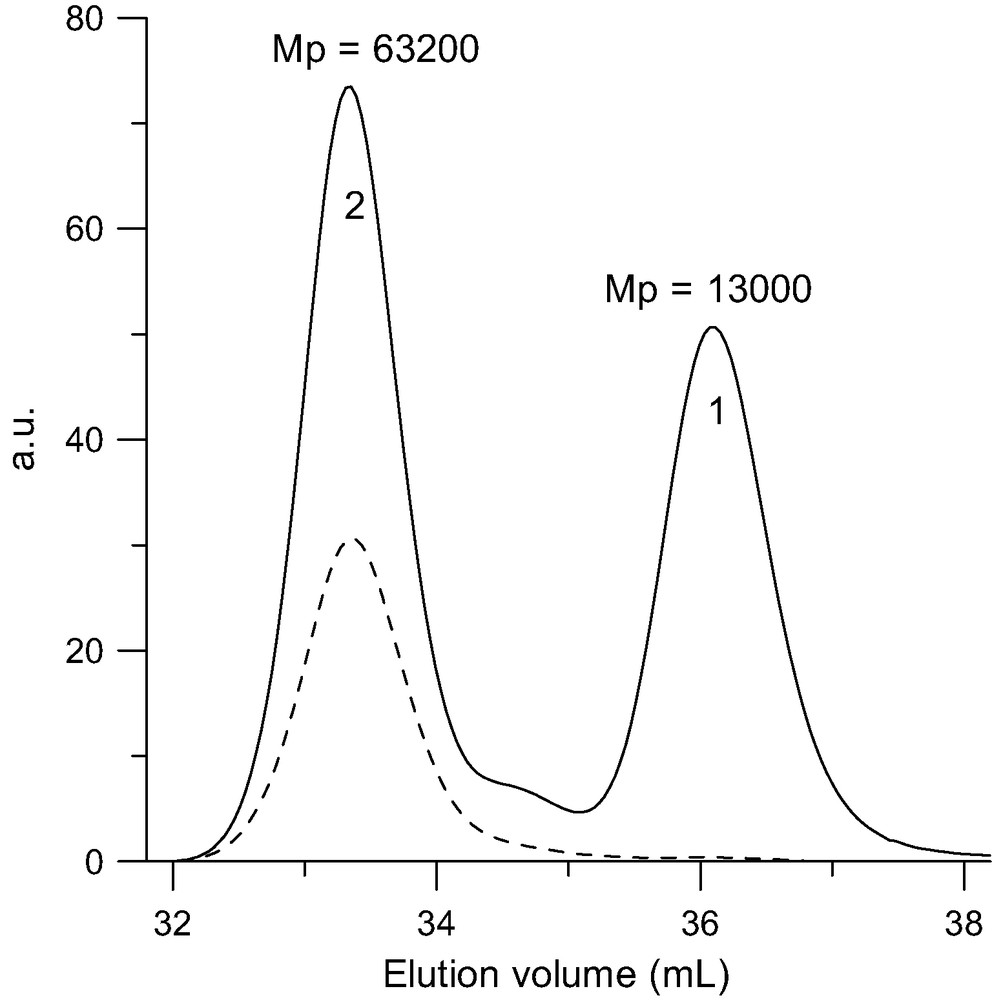
SEC analysis of the hexa-adducts (2) (MwLS = 83 000 g/mol) upon reacting 10 PSLi (1) (Mw = 14 000 g/mol) with C60. Solid line: RI trace. Doted line: UV trace at 320 nm where only the fullerene-containing products are detected.
SEC and LS characterization of some six-arm polystyrene stars with a C60 core and their ‘parent’ PS–Li. As only LS is able to give the actual value of the molar mass of a branched polymer, the experimental functionality of the stars is derived from MwLSstar/MwLSarm
| Sample | SEC-RI detector |
| Star functionality | |
| Mw | Mw/Mn | |||
| Aarm | 360 000 | 1.16 | 347 000 | |
| Astar | 1 290 000 | 1.09 | 2 045 000 | 5.9 |
| Barm | 89 700 | 1.05 | 88 000 | |
| Bstar | 396 000 | 1.09 | 536 000 | 6.1 |
| Carm | 35 300 | 1.04 | 32 500 | |
| Cstar | 142 800 | 1.06 | 192 000 | 5.9 |
| Darm | 13 800 | 1.09 | 14 000 | |
| Dstar | 64 200 | 1.08 | 83 000 | 5.9 |
| Earm | 5 600 | 1.04 | – | |
| Estar | 26 400 | 1.08 | 33 000 | 5.9 |
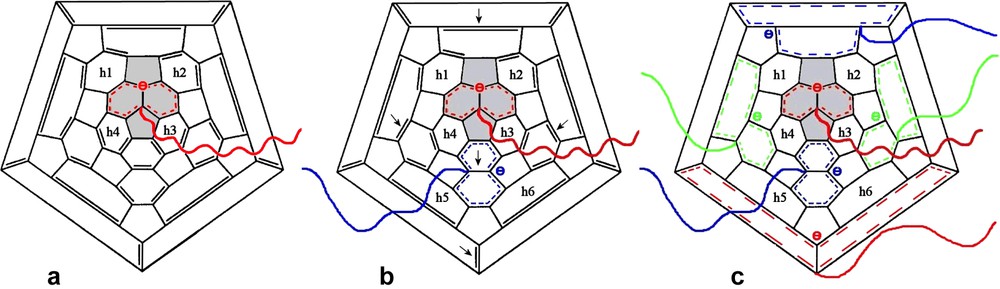
Delocalization of the carbanions introduced on the C60 upon addition of PSLi. Explanation of the upper limit of six grafted chains (see text).
SEC analysis of the six-arm block-copolymer star (PI-b-PS)6C60 (MwLS = 220 000 g/mol) upon reacting seven PI-b-PSLi (MwLS = 35 000 g/mol) with C60. Solid line: RI trace for (PI-b-PS)6C60 (2) and excess of PI-b-PSLi (3). Doted line: UV trace at 320 nm. Dot-point line: RI trace for the PI block (MwLS = 27 000 g/mol).
As already mentioned above, stoichiometric control allows to prepare tri-, tetra- and penta-adducts. The upper limit of the number of grafts being 6, it becomes possible to add, respectively, 3, 2 or 1 additional PS or PI arms. That has been used to prepare asymmetric stars (PSa)6–nC60(PSb)n or mikto-arm stars (PS)6–nC60(PI)n by reacting such adducts of lower functionality with an excess of PSLi or polyisoprenyllithium (PILi) of a different molar mass [13]. The same approach was used to add five additional PS or PI chains on a mono-adduct PS–C60 prepared by reacting the fullerene with an azide-terminated polystyrene [14].
Another interest of this grafting route comes from the fact that anionic polymerization allows to prepare ‘living’ block-copolymers that can then be attached to the fullerene. An example of the SEC characterization of such a six-arm star where the arms consist of a polyisoprene-b-polystyrene block copolymer is given in Fig. 3. The copolymer arms were grafted to the C60 core through their PS part to avoid stability problems (see below). The organization of these block-copolymer stars in the solid state, which leads to a spatial ordering of the fullerene cores, is under investigation.
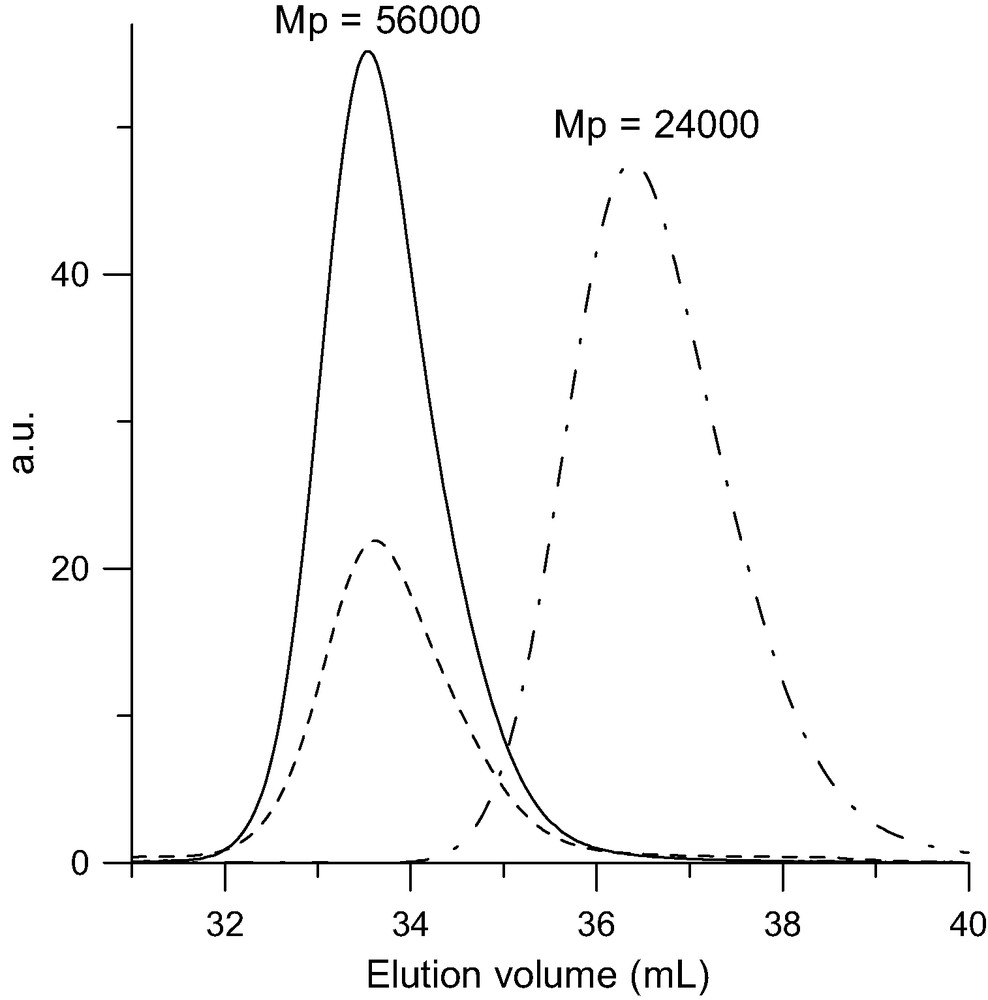
SEC analysis of the di-adduct (Mw = 62 000 g/mol) formed upon reacting one PSBr with one C60 in toluene at 100–110 °C for 50 h in the presence of CuBr/bipyridine. Dot-point line: RI trace of the pristine PSBr (Mw = 27 000 g/mol). Solid line: RI trace of di-adduct. Doted line: UV trace at 320 nm.
2.1.2 In polar solvents
The C60 being one of the best electron acceptors, the mechanism of the reaction between this fullerene and a ‘living’ anionic polymer becomes more complicated in a polar solvent like THF where electron transfer can occur. Indeed, using UV/Vis/NIR spectroscopy we could show that reacting C60 with increased amounts of a ‘living’ PS–K+ in THF leads first to the quantitative formation of the mono-anion C601–K+ and then to the di-anion C602–(K+)2 through electron transfer from the carbanion. Only after all the fullerenes are converted to di-anions, addition of up to four chains is observed [9,15] as illustrated in Scheme 3. The electron transfer step can be completely separated from the addition step by reacting PS–K+ directly with C602–(K+)2 prepared separately. That allows preparing tetra-adducts where the C60 core bears six negative charges [9]. Networks with C60 knots could also be prepared by reacting α,ω-dicarbanionic ‘living’ (PS)2–(K+)2 chains with di-anions C602–(K+)2 [16]. Electron transfer from the carbanion to the fullerene occurs already when only a few % of polar solvents are added to a non-polar one and even electron transfer from oxanions was observed [17]. That limits the controlled addition of ‘living’ carbanionic polymer chains onto C60 to the ones that can be prepared in non-polar solvents.
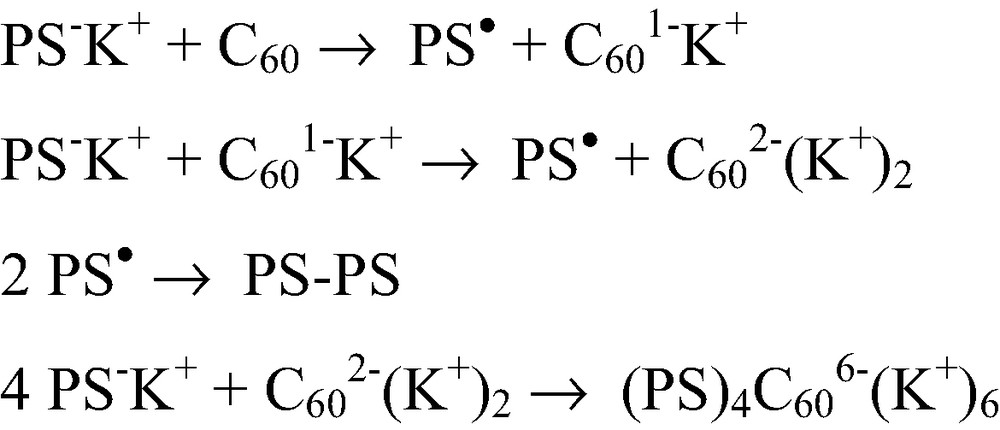
Reaction mechanism between C60 and a ‘living’ carbanionic polymer in THF.
2.1.3 ‘Living’ hexa-adducts (PS)6C606–(Li+)6 as initiators for anionic polymerization
If six carbanions are located on the fullerene core their delocalization is restricted and the negative charges become reactive enough to open the double bond of a vinyl monomer such as styrene, butadiene or isoprene [18]. But, as can be seen in Scheme 4, if one of the six carbanions adds to a monomer the negative charge leaves the fullerene and the remaining charges become more delocalized. And it is well known that the reactivity of a carbanion decreases if the delocalization of the charge increases. We were able to demonstrate that the reactivity of the five remaining carbanions is sufficiently decreased so that they are not longer able to open the double bond of non-polar monomers like styrene, butadiene or isoprene. So, if one of these three monomers is added to a ‘living’ hexa-adduct, only one chain is growing out from the fullerene core. As this additional chain is produced by anionic polymerization its polydispersity stays low and its molar mass can be controlled. We have taken advantage of this very specific initiation mechanism, never encountered before in the in–out synthesis of stars by anionic polymerization, to prepare ‘palm-tree’-like architectures where the C60 core bears six PS chains of the same length and an additional seventh chain of a different length or/and a different chemical nature (polyisoprene for example) [19]. Furthermore, as the out-growing chain bears a terminal carbanion it is possible to specifically couple two ‘palm-trees’ with for example dibromoparaxylene to form ‘dumbbell’-type architectures where two six-arm PS stars with a fullerene core are linked together by a PS or a polyisoprene chain [19,20]. A schematic representation of these ‘palm-tree’ and ‘dumbbell’ architectures is given in Scheme 5.

Consequences of the addition of a vinyl monomer onto one of the six carbanions of a ‘living’ hexa-adduct on the delocalization of the negative charges.

Schematic representation of ‘palm-tree’- and ‘dumbbell’- like architectures.
3 Grafting of polystyrene chains onto C60 by an atom transfer radical addition
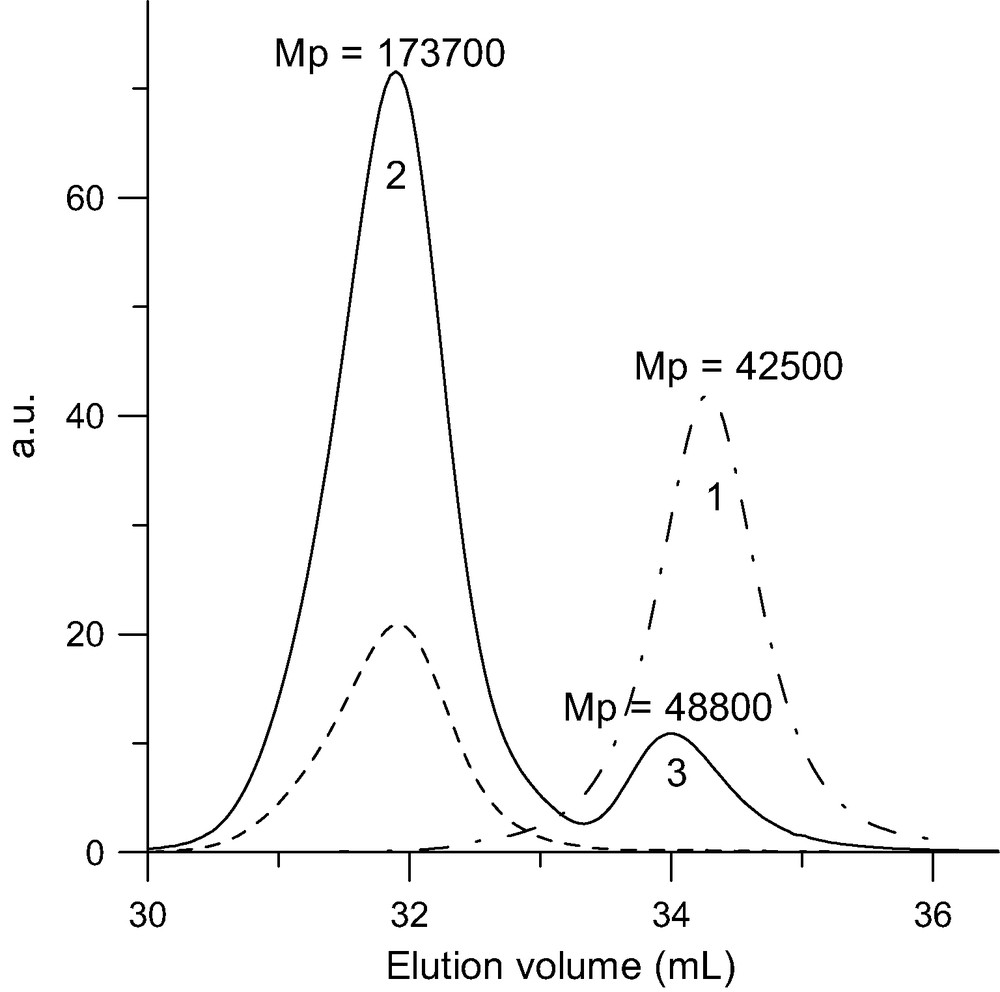
The C60 molecule is known to be a good radical acceptor [21] and grafting of radicals onto this fullerene was one of the first addition reaction reported in [22]. A simple radical copolymerization of C60 with a vinyl monomer such as styrene or methyl methacrylate allows to covalently bond fullerenes on the polymer [23–31]. But the products obtained are ill-defined as neither the number nor the length of grafts is controlled. Recent progress in ‘controlled’ radical polymerization allows preparing polymer chains with a terminal function that can be converted to a radical. That opens the possibility to graft macro-radicals of controlled molar mass and low polydispersity onto the C60 [32–37]. Di-adducts (PS)2C60 have been prepared by reacting the fullerene with PS chains terminated with a stable counter radical [33,34]. A first report on the grafting onto C60 of low molar masses bromine terminated polystyrene (PSBr) and polymethylmethacrylate (PMMABr) prepared by an atom transfer radical polymerization (ATRP) via an atom transfer radical addition (ATRA) concluded that mono-adducts are formed [36]. But, this conclusion is questionable, as a more convincing interpretation of the presented SEC data would in fact support the formation of di-adducts. Our own work on the addition of PSBr onto C60 by ATRA [37,38] showed that no mono-adducts are obtained even if the C60 is used in a 10-fold excess and only the grafting of an even number of chains is observed. Pure di-adducts are produced if the ratio PSBr/C60 is kept lower than 2 and we were able to demonstrate for the first time that pure tetra-adducts can also be obtained if the excess of PSBr over C60 is larger than 4. In this latter case, polymer fractionation was applied to remove the excess of ungrafted chains. An example of the SEC traces for a di- and a tetra-adduct along with their ‘parent’ PSBr is given, respectively, in Figs. 3 and 4. Table 2 presents the SEC and LS characterization of some two- and four-arm polystyrene stars with a C60 core along with their ‘parent’ PS–Br in order to allow determining the experimental functionality of the stars. The mechanism responsible for the grafting of only even number of chains onto C60 through this chemistry is given in Scheme 6. The addition of even number of chains results from the fact that any bond between the fullerene and an atom A is easier to break than a ‘normal’ C–A bond. After a first addition of a PSBr onto C60 through ATRA, a C60–Br bond is formed (Scheme 6a). But, as this latter is easier to break as the terminal PS–Br bond, the concentration of PS–C60● in the radical form is always large enough so that any macro-radical PS● formed will preferentially recombine with the radical present on the fullerene. From the UV–vis spectrum of di-adducts it was possible to deduce that the addition of the two arms occurs in 1,4 position on the same hexagon and leads to the opening of only one double bond [38]. An additional proof of this mechanism comes from the absence of Br in the stars as shown by elemental analysis of low-molar mass adducts [38]. The same behavior accounts for the absence of tri-adducts and the direct formation of tetra-adducts if the ratio PSBr/C60 becomes higher than 2. This chemistry allows to prepare asymmetric stars (PSa)2C60(PSb)2 or mikto-arm stars (PSa)2C60(PI)2 in a very simple way. In a first step a PSaBr is reacted with C60 in a ratio PSaBr/C60 lower than 2 and the excess of fullerene removed from the di-adduct using a polar solvent where C60 is insoluble. The di-adduct is then reacted with an excess of PSbBr of a different molar mass or with a Br terminated polyisoprene under ATRA conditions to obtain the asymmetric or mikto-arm tetra-adduct [13]. Furthermore, as the limit of the chains grafted by using ‘living’ anionic polymers is 6, it should be possible to graft additional chains onto the C60 core of di- and tetra-adducts prepared through ATRA. Work is in progress in this direction.
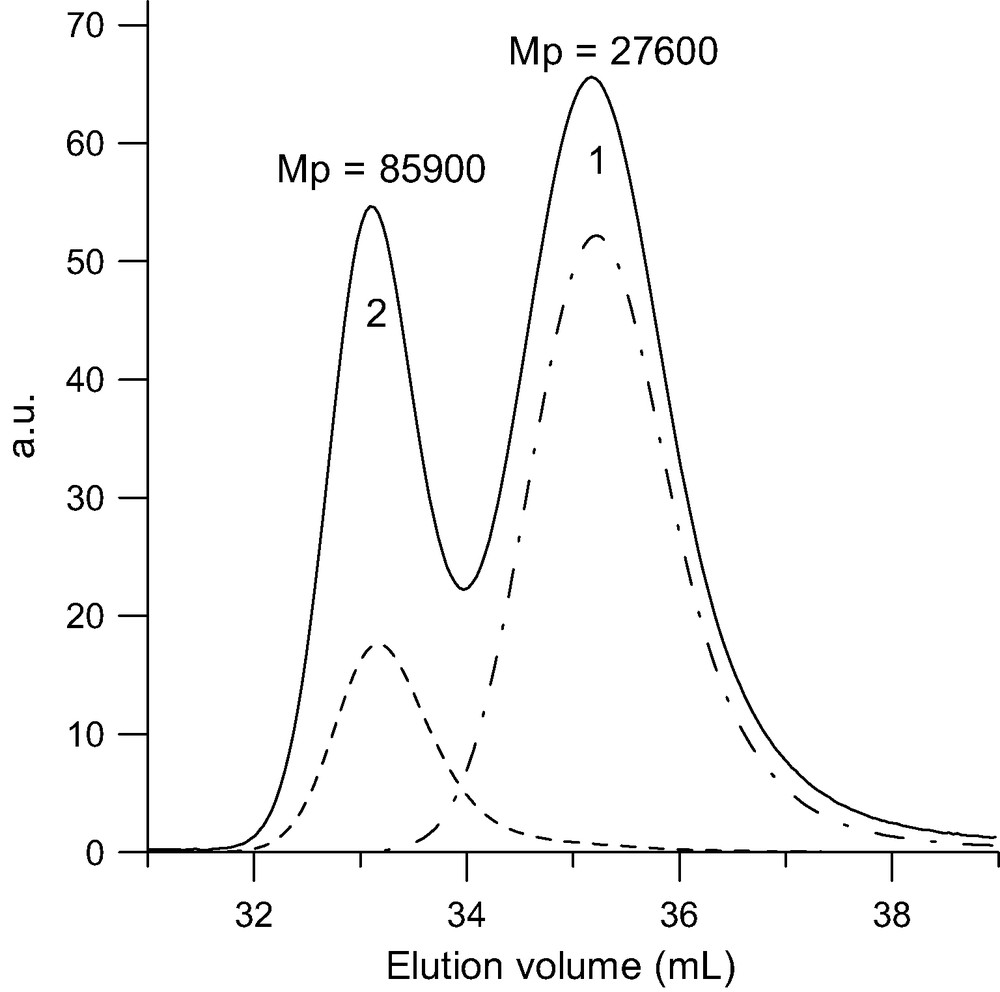
SEC analysis of the tetra-adduct (MwLS = 101 000 g/mol) formed upon reacting a 10-fold excess of PSBr (MwLS = 26 000 g/mol) with C60 in toluene at 100–110 °C for 65 h in the presence of CuBr/bipyridine. Dot-point line: RI trace of the pristine PSBr. Solid line: RI trace of tetra-adduct (2) and excess of PSBr (1). Doted line: UV trace at 320 nm.
SEC and LS characterization of some two- and four-arm polystyrene stars with a C60 core and their ‘parent’ PS–Br. The experimental functionality of two- and four-arm stars is derived, respectively, from MwSECstar/MwSECarm and MwLSstar/MwLSarm
| Sample | SEC-RI detector |
| Star functionality | |
| Mw | Mw/Mn | |||
| Farm | 29 000 | 1.25 | – | |
| Fstar | 67 000 | 1.20 | – | 2.3 |
| Garm | 42 000 | 1.18 | – | |
| Gstar | 88 000 | 1.21 | – | 2.1 |
| Harm | 28 000 | 1.20 | 26 000 | |
| Hstar | 87 000 | 1.17 | 101 000 | 3.9 |
| Iarm | 49 500 | 1.24 | 42 000 | |
| Istar | 135 000 | 1.09 | 160 000 | 3.8 |
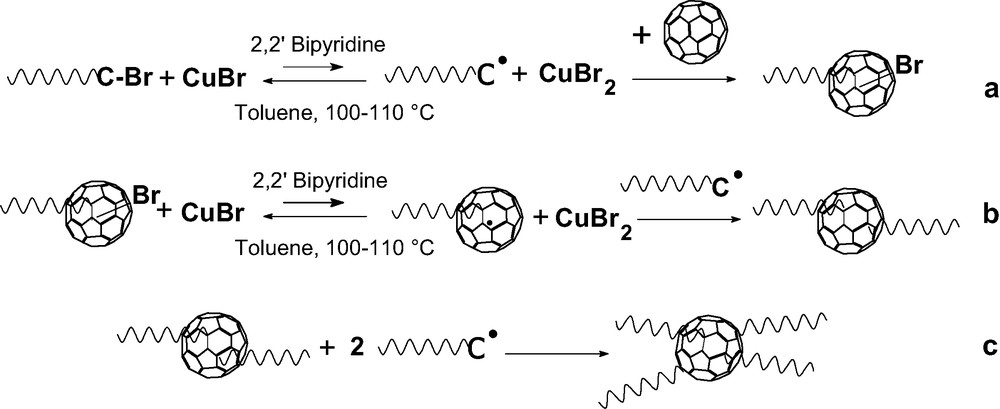
Addition mechanism of PSBr chains onto C60 through an ATRA.
4 Thermal stability of (PS)xC60 stars
Due to the presence of a C60 core these polystyrene stars present interesting photophysical characteristics [39] such as optical limiting [40] and non-linear optical properties [41,42]. But, for applications in these fields the thermal stability of these materials is a crucial issue. For this reason we studied the thermal stability both in bulk using thermal gravimetric analysis (TGA) and in solution by following the evolution of the molar mass of the stars as a function of heating time by SEC. An example of the TGA analysis of a (PS5400)6C60 is given in Fig. 5 and compared to that of a linear polystyrene. The thermal decomposition of the star with a C60 core starts at about 100 °C before that of linear PS and, from the derivative of the variation of weight with temperature, it appears that the thermal decomposition occurs in two steps. Analysis of the volatile products by mass spectroscopy has shown that styrene is produced all over the decomposition process, but that all the C60 comes out during the low temperature step [43,44]. These observations are in agreement with the well known thermal decomposition of PS through a radical initiated depolymerization and they imply that all the links between the fullerene and the arms are broken in the low temperature step. That supposes that the PS–C60 bond is weaker than the C–C bonds in the polystyrene chain. This point has been confirmed by the thermal stability study in solution. Indeed, heating a toluene solution of a (PS)6C60 at around 100 °C for several days leads to a progressive decrease of the molar mass of the star along with a quantitative release of the PS arms and no changes in the molar mass of these latter [45]. In addition, this study showed that the thermal stability of a PS star with a C60 core increases if the number of arms decreases [46]. This is illustrated in Fig. 6 where the progressive release of the PS arms upon heating at 160 °C a 2-methylnaphtalene solution of a four-arm star is presented.

Thermal degradation of a (PS5400)6C60 star and of a linear polystyrene measured by TGA.
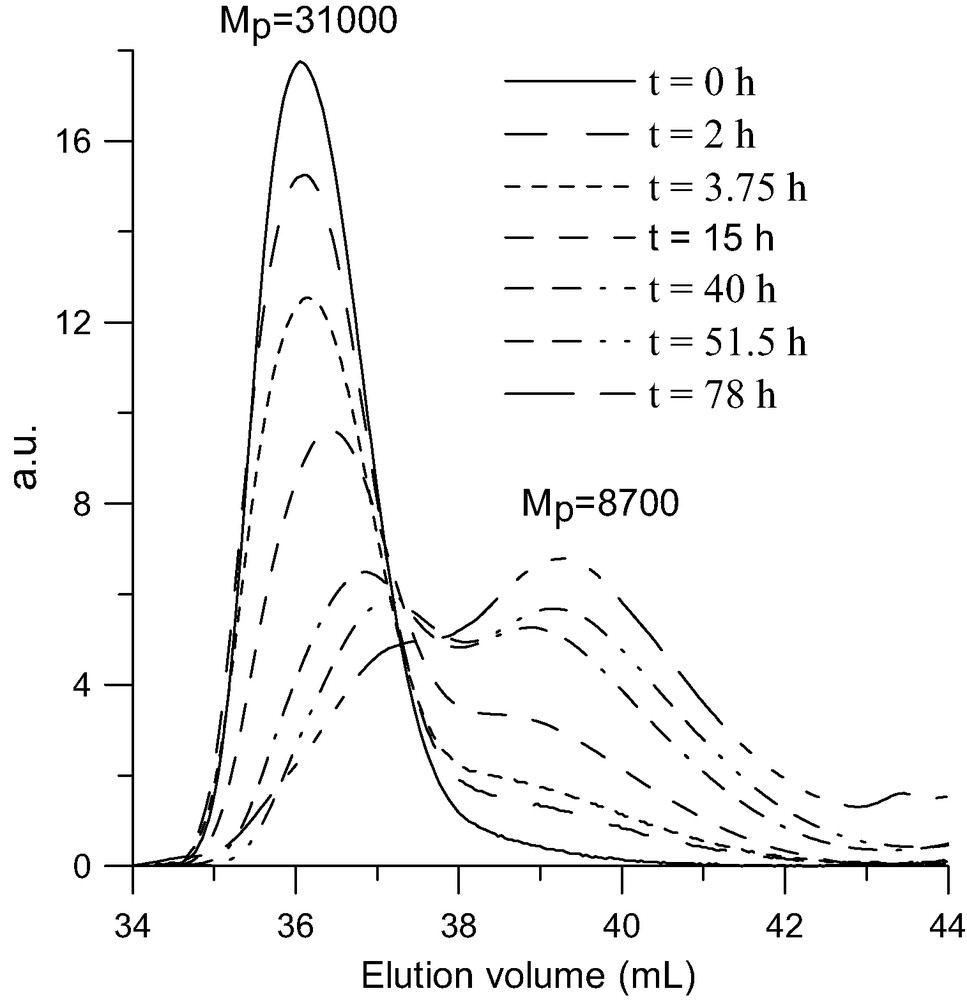
SEC analysis of thermal decomposition of a (PS8800)4C60 star (MwLS = 34 500 g/mol) at 160 °C in solution in 2-methylnaphtalene. RI detector.
So, having demonstrated that it is indeed the bond between the PS arm and the fullerene that breaks first, the thermal decomposition in two steps (Fig. 5) can be rationalized as shown in Scheme 7. At around 350 °C the PS–C60 bonds break, releasing the fullerene and creating on the end of the chain a radical that starts the depolymerization process (low temperature step). But, at this temperature the depolymerization is not complete and it stops by dismutation or recombination of the radicals leaving behind a part of the PS chain. This latter including not longer the “weak” arm-fullerene bond stays then stable up to the normal depolymerization temperature of polystyrene (high-temperature step). From the weight loss in the low temperature step measured by TGA on stars with arms of various molar masses [44] we could conclude that, whatever is the molar mass of the arm, around 10–15 monomer units are released after the cleavage of a PS–C60 bond. A closer look at the high temperature step seems to indicate some decreased of the thermal stability of the remaining part of the PS arms as compared to linear PS. That can result from the formation of the somewhat weaker head to head bonds by recombination of the PS● radicals generated in the low-temperature step.
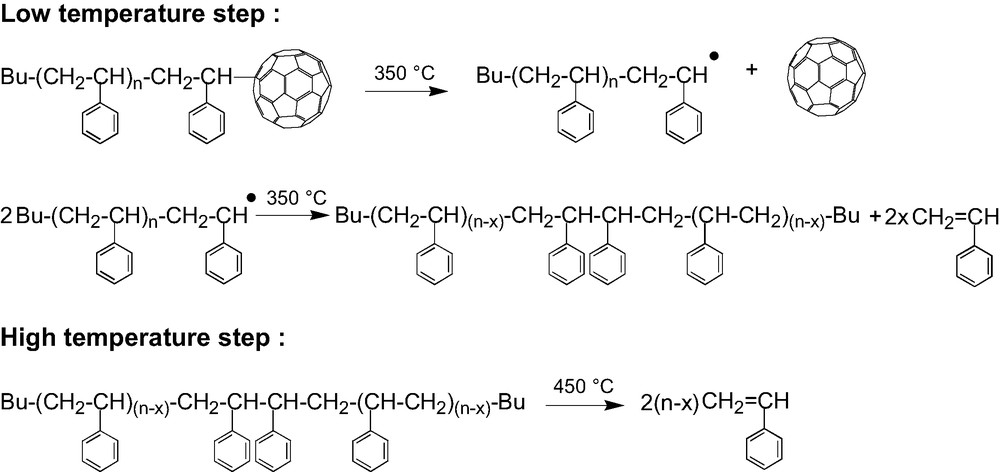
Thermal degradation mechanism for (PS)6C60.
5 Nanoscale organization of C60 in a polymer matrix by self-assembly of block-copolymers
The possibility to synthesize well defined homopolystyrene (hPS) stars C60(PS)f (f = 2, 4 and 6) as well as block-copolymer stars C60(PS-b-PI)6 opens new and interesting perspectives for the nanoscale organization of C60 in a polymer matrix. It is well known that block-copolymers made of two incompatible sequences self-organize in the solid state in order to minimize the interface between the incompatible domains. Depending on the respective molar masses of the blocs, various morphologies (spheres, hexagonal array of cylinders, gyroïd, lamellae) are formed [47]. Furthermore, it has been shown that the addition of hPS to a diblock copolymer PS-b-PI can result in the selective solubilization of the hPS into the PS domains of the microphase separated structure of the diblock copolymer [48]. Following the same approach, we have demonstrated that selective solubilization of hPS stars C60(PS)x (x = 2, 4 and 6) in the PS lamellae of a symmetric PS-b-PI copolymer allows to create a periodic nanoscale organization of C60 in a copolymer matrix (schematized in Fig. 7) with a typical lamellar periodicity D in the range 20–100 nm [49]. As an example, the swollen lamellar morphology observed by transmission electron microscopy (TEM) for blends of a PS30000-b-PI30000 copolymer with 40 wt.% of a C60(PS)6 star with Mw = 18 000 g/mol is given in Fig. 7.
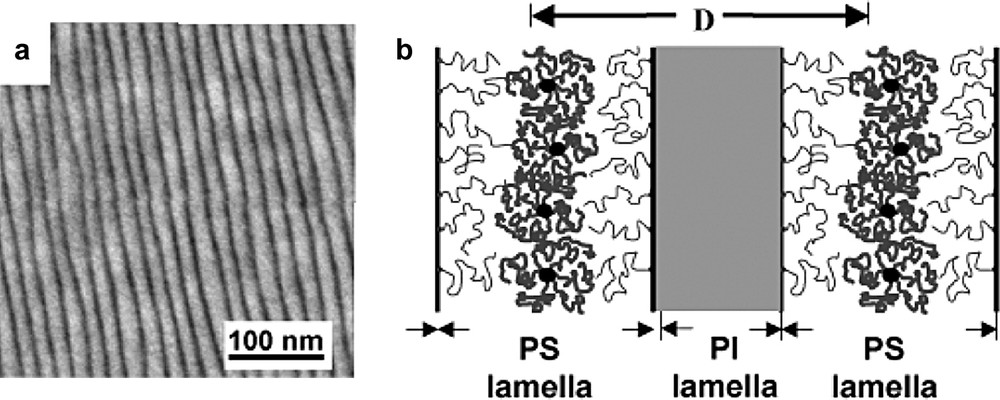
(a) TEM micrograph showing the swollen lamellar morphology of a PS30000-b-PI30000 copolymer blended with 40 wt.% of a C60(PS)6 star with Mw = 18 000 g/mol. (b) Schematic view of the localized solubilization of the C60(PS)6 stars in the PS lamellae of the symmetric block copolymer. Note that the PI lamella appear dark (OsO4 staining).
We have also started to take profit of the unique control in the synthesis of star copolymers e.g. C60(PS-b-PI)6 with a low polydispersity (Mn/Mw = 1.06) to achieve a nanoscale organization of C60 [50]. In contrast to the first approach involving blends of a structuring copolymer matrix and C60(PS)f stars, we obtain directly the nanoscale organization via self-assembly of the copolymer chains that are grafted onto the C60 core. In Fig. 8, we show the typical lamellar morphology obtained for a C60(PS25000-b-PI25000)6 star block copolymer. This approach is expected to lead to a higher degree of localization of the C60 cores towards the middle of the PS lamellae. Furthermore, by changing the volume fraction of the blocks, one can obtain all the typical microphase separated morphologies e.g. hexagonal packing of cylinders and more interestingly the gyroid structure that involves two interpenetrated and continuous 3D networks of C60.
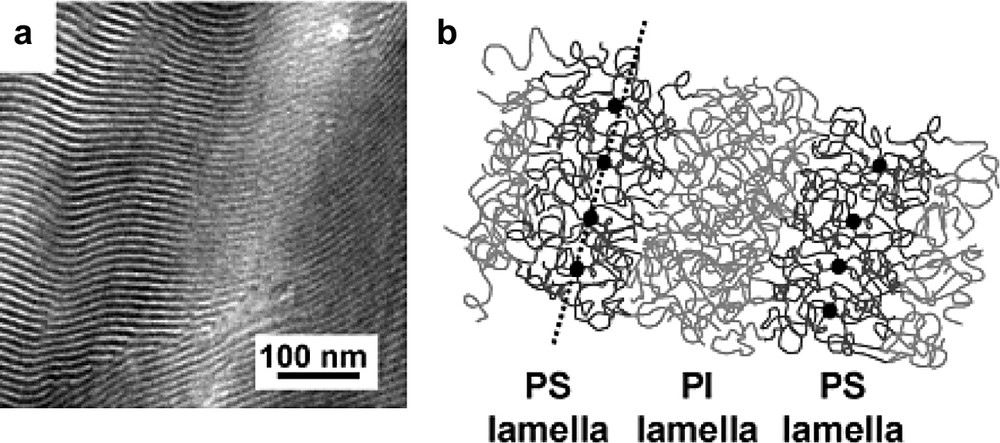
(a) TEM micrograph showing the lamellar microstructure of a C60(PS25000-b-PI25000)6 star block copolymer. (b) Schematic representation of the nanoscale organization of the C60 cores via self-assembly of the PS-b-PI arms of the stars.
6 Conclusion
Well-defined stars with a fullerene core and arms of defined molar masses and low polydispersities can be prepared through addition onto C60 of ‘living’ anionic polymers or by grafting macro-radicals generated using an atom transfer mechanism. A good knowledge of the peculiar addition mechanisms resulting from the specific chemical reactivity of the C60 allows to perfectly control the functionality of the stars (i.e. the number of opened double bonds) and so the electronic properties of the incorporated fullerene. The delocalization of the carbanions, introduced on the C60 upon addition of ‘living’ polymers, on a single conjugated molecule accounts not only for the upper limit of six grafts, but allows to produce more sophisticated architectures such as ‘palm-trees’ or ‘dumbbells’. All these compounds display a decreased thermal stability attributed to the presence of covalent bonds between the fullerene core and the arms. Indeed, this C60-arm bond has been found to be less stable than the C–C bonds in the polymer chains. But, even if the thermal stability of (PS)xC60 stars is decreased as compared to normal polystyrene, these materials are stable enough to survive classical polymer processing. Beside their interest in NLO, the presence of grafted polymer chains on the fullerene allows to achieve spatial organization of C60 in a structuring block-copolymer matrix.
Acknowledgements
We would like to thank O. Gavat, C. Foussat, Y. Guilbert, R. Meens and A. Rameau for technical support.