

1 Introduction
Industrial development and the strong increase of transportation has lead to more and more environmental pollution [1]. Consequently, specific and rigorous standards for gasoline and diesel fuels must be applied. For instance, the sulfur content in gasoline and diesel fuels is continuously reduced by regulations to lower and lower levels. In Europe and USA, new sulfur content limits of about 30–50 ppm in gasoline and diesel are required. Moreover, these sulfur levels are expected to be lowered to 10 ppm by the end of this decade and “zero sulfur” emissions are targeted in the future [2–4]. In order to face these demanding environmental standards, the development of more active hydrodesulfurization (HDS) catalysts seems to be the more promising way to improve fuels quality without negative impact on capital investment (compatibility of the HDS catalysts with the current HDS infrastructures) [1]. In order to achieve this goal, many approaches were attempted, like changing the active component, varying the preparation method and changing the support.
In industrial processes, the hydrotreating catalysts are generally composed of a molybdenum sulfide phase “MoS2” or a tungsten sulfide phase “WS2” promoted by cobalt or nickel and usually supported on gamma alumina (γ-Al2O3) [5–12]. The use of γ-Al2O3 as a support has to be ascribed to its mechanical properties, to a relatively low cost [13] and to its ability to provide high dispersion of the active metal phase [8]. Previously, it was assumed by Candia et al. that, upon increasing the sulfiding temperature, the nature of the CoMoS phase structure changes from a low temperature form “CoMoS I phase” to a high temperature form “CoMoS II phase” [6]. The type I phase, i.e. “CoMoS I”, would be incompletely sulfided, highly dispersed and strongly bonded to the surface of alumina via Mo–O–Al anchoring points [14] and such Mo–O–Al linkages induce lower HDS activities [15]. In contrast, “CoMoS II” phase is supposed to be completely sulfided, highly stacked and interact weakly with alumina via Van der Waals interactions without Mo–O–Al anchoring points [6,14]. The pure “CoMoS II phase” presented a much higher activity (twice) than the “CoMoS I phase”. This problem of interactions between the active phase and alumina has urged numerous researches to develop and evaluate other supports. Thus, by changing the support (carbon, titania or zirconia), it was shown that the resulting unpromoted catalysts presented different and higher catalytic activities than those supported on alumina [16–18]. For instance, Vrinat et al. and Muralidhar et al. previously showed that unpromoted molybdenum sulfide “MoS2” catalysts prepared on TiO2 were 3–5 times more active than the same formulation supported on γ-Al2O3 [16,19]. Nevertheless, the preparation of promoted catalysts by successive impregnation of ammonium heptamolybdate and cobalt nitrate gave different synergetic effects depending on the support. For TiO2 promoted catalysts for instance, the synergetic effects were always lower than those observed on γ-Al2O3. As suggested in the literature, this phenomenon is probably due to a phase segregation between molybdenum sulfide and the promoter: parasitic phases are formed leading to low catalytic activities and consequently limiting the interest of changing the support [16,19,20]. Thus, the main objective of this work was to study the support effect by using a non-classical synthesis method: the addition of the promoter directly on the molybdenum sulfide phase in order to minimize the interaction between the Co and the support and to optimize CoMoS phase formation. For that purpose, the promotion of the solids MoS2/γ-Al2O3, MoS2/TiO2 and MoS2/SiO2, containing the same loading of molybdenum per surface unit was made using cobalt nitrate or cobalt acetylacetonate. These catalysts were compared with the solids obtained by a classical preparation method (co-impregnation of Co and Mo) and characterized by elemental analyses, high-resolution electron microscopy (HREM) and X-ray photoelectron spectroscopy (XPS).
The catalytic activities were evaluated in the hydrodesulfurization reaction of thiophene and the results are discussed.
2 Experimental procedures
2.1 General procedure
The supports used in the present study are: TiO2 (SBET = 154 m2 g−1, ØPores = 7.8 nm, Vp = 0.34 cm3 g−1), SiO2 (SBET = 305 m2 g−1, ØPores = 11.8 nm, Vp = 1.13 cm3 g−1) and γ-Al2O3 (SBET = 302 m2 g−1, ØPores = 8.0 nm, Vp = 0.77 cm3 g−1). The supports were crushed and sieved to particles between 80 and 125 μm for the preparation of catalysts. The measurements of the textural properties were made by physical adsorption of nitrogen (adsorption–desorption isotherms), which allowed the determination of the porous diameters, porous volumes and specific surface areas of the different supports and prepared catalysts. For these experiments, the samples were desorbed in vacuum for 3 h at 573 K.
2.2 Preparation of supported molybdenum oxide
Due to the large differences between the surface areas of the supports, and in order to evaluate the intrinsic potential of the supports, 2.5 atoms of molybdenum per square nanometer (2.5 atoms of Mo nm−2) have been loaded on each support using the incipient wetness impregnation technique with an aqueous solution of ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24·4H2O). After impregnation, the catalyst was dried at 383 K overnight, and finally calcined at 773 K under airflow for 5 h.
2.3 Impregnation of Co(NO3)2·6H2O on supported molybdenum oxide
The impregnation of cobalt on supported molybdenum oxide was carried out using an aqueous solution of cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) in order to obtain an atomic ratio Co/(Co + Mo) = 0.3. The solid was then dried at 383 K overnight, and finally calcined at 773 K under airflow for 5 h. Before thiophene HDS experiments, the catalysts were sulfided at atmospheric pressure with a H2–H2S (15%) mixture for 4 h at 673 K. These catalysts are referred as CoMoS–O/support.
2.4 Impregnation of Co(NO3)2·6H2O on supported molybdenum sulfide
The same procedure as described above was used, but in this case the addition of cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) was realized on the presulfided molybdenum catalyst (MoS2). Thus, after the impregnation of molybdenum precursor using the incipient wetness impregnation technique, the catalyst was dried, calcined and sulfided, as described previously, in order to obtain supported MoS2. Then these supported MoS2 solids were impregnated with an aqueous solution of the promoter Co(NO3)2·6H2O (Co/(Co + Mo) = 0.3). The resulting catalysts were dried again, calcined, resulfided and referred as CoMoS–S/support.
2.5 Impregnation of Co(acac)2·nH2O on supported molybdenum sulfide
To promote the samples with Co(acac)2·nH2O, the refluxing method was used because of the low solubility of the acetylacetonate promoter in water. In this case, the required amount of acetylacetonate complex (Co/(Co + Mo) = 0.3) was dissolved in methanol and then added to supported MoS2. The suspension was heated at boiling temperature (338 K) for 4 h under argon. The resulting solids were dried at 353 K and resuling, as described previously, without further calcination. They are referred as CoMoS–acac/support.
2.6 Catalysts characterizations
Elemental analyses of Mo and Co content were performed using plasma coupled atomic emission spectroscopy (AES–ICP).
High-resolution electron microscopy (HREM) analyses of unpromoted catalysts were performed using JEOL 2010 (200 kV) microscope. As the microscope is not equipped with a pretreatment chamber, samples to be analyzed by HREM were sulfided beforehand, and kept under argon atmosphere. Supported MoS2 catalysts were crushed then dispersed in pure ethanol by ultrasounds. Then a drop of the homogenized suspension was deposited on a copper grid with holes of 200 mesh, covered with a carbon film with holes.
X-ray photoelectron spectroscopy (XPS) analyses of unpromoted catalysts were carried out under an inert atmosphere using AXIS Ultra DLD spectrometer equipped with a Delay Line Detector. XPS measurements were taken with a monochromatized aluminum source (1486.6 eV) and a correction of the binding energy was carried out taking as reference, according to the support, Al 2p (74.1 eV) or C 1s (284.6 eV). The data processing (acquisition, substraction of the background and decomposition) was carried out using the software XPS processing.
2.7 Catalytic activities measurements
Thiophene HDS measurements were carried out in a fixed-bed flow microreactor, at atmospheric pressure, with a thiophene partial pressure of 21.4 Torr and in the reaction temperature range between 573 K and 613 K. The specific rate was determined after 16 h time on stream using the relation As = [Q·ln(1/(1 − τ))]/m in which As represents the specific rate (mol s−1 g−1), Q the molar flow rate of reactant (mol of thiophene s−1), τ the conversion and m the weight of catalyst (g).
3 Results
3.1 Compositions and textural characteristics of supports and catalysts
Specific surface area and porosity are very important parameters in the field of heterogeneous catalysis. These properties control the diffusion phenomena in the solids and the catalytic reaction selectivities [21]. The surface areas, average pore volumes and pore diameters of the supports as well as the surface areas and elemental analyses of the different promoted and unpromoted catalysts are presented in Table 1. The high surface areas of gamma alumina and silica contrast with the relatively low surface area of titania. The Mo catalysts on γ-Al2O3 and SiO2, as well as, the CoMoS catalysts on γ-Al2O3, TiO2 and SiO2, exhibited, as expected, smaller surface areas than the corresponding supports due to the increased catalyst density and to the support pore blockage by metal particles. Mo elemental analyses of the unpromoted catalysts “MoS2” showed that, on each support, a molybdenum surface coverage of approximately 2.5 Mo atoms per square nanometer had been obtained. Moreover, Co elemental analyses of the different promoted catalysts “CoMoS” varies between 1.2 and 3.5%, which corresponds to an atomic Co/(Co + Mo) ratio of about 0.3, a value often required to get good catalytic activities in HDS [22].
Composition and textural characteristics of the supports and catalytic systems.
| Material | SBETa (m2 g−1) | PVb (cm3 g−1) | PDc (nm) | Mod (wt%) | Cod (wt%) | Atomic ratio Co/(Co + Mo)d |
| TiO2 | 153 | 0.35 | 7.8 | – | – | – |
| SiO2 | 305 | 1.13 | 11.8 | – | – | – |
| γ-Al2O3 | 302 | 0.77 | 8.0 | – | – | – |
| MoS2/TiO2 | 156 | – | – | 5.97 | – | – |
| MoS2/SiO2 | 266 | – | – | 12.0 | – | – |
| MoS2/γ-Al2O3 | 245 | – | – | 11.2 | – | – |
| CoMoS–O/TiO2 | 123 | – | – | 5.97 | 1.48 | 0.29 |
| CoMoS–S/TiO2 | 126 | – | – | 5.97 | 1.21 | 0.26 |
| CoMoS–acac/TiO2 | 128 | – | – | 5.97 | 1.26 | 0.27 |
| CoMoS–O/SiO2 | 261 | – | – | 12.0 | 3.48 | 0.31 |
| CoMoS–S/SiO2 | 259 | – | – | 12.0 | 2.97 | 0.29 |
| CoMoS–acac/SiO2 | 260 | – | – | 12.0 | 2.64 | 0.26 |
| CoMoS–O/γ-Al2O3 | 235 | – | – | 11.2 | 2.68 | 0.28 |
| CoMoS–S/γ-Al2O3 | 240 | – | – | 11.2 | 2.31 | 0.26 |
| CoMoS–acac/γ-Al2O3 | 230 | – | – | 11.2 | 2.14 | 0.25 |
a Surface area determined by BET method.
b Pore volume average determined by t method.
c Pore diameter average determined by BJH method.
d Elemental analyses' experimental values.
3.2 Thiophene HDS activities of unpromoted molybdenum catalysts
Supported unpromoted molybdenum catalysts were evaluated in thiophene HDS. The values of specific activities (per gram of catalyst and per second), as well as the values of intrinsic activities (thiophene molecule per Mo atom and per second), are reported in Table 2. The higher specific and intrinsic activities have been found with the MoS2 catalyst supported on titania as expected.
Catalytic activities of supported unpromoted molybdenum catalysts.
| Catalyst | 573 K | 593 K | 613 K | ||||||
| τ | V | Vi | τ | V | Vi | τ | V | Vi | |
| MoS2/TiO2 | 4.2 | 41 | 6.6 | 7.0 | 58 | 9.4 | 11.5 | 75 | 12.2 |
| MoS2/SiO2 | 4.1 | 19 | 1.6 | 7.0 | 33 | 2.6 | 12.0 | 58 | 4.6 |
| MoS2/γ-Al2O3 | 3.9 | 20 | 1.7 | 6.3 | 32 | 2.7 | 10.1 | 52 | 4.4 |
3.3 Thiophene HDS activities of promoted molybdenum catalysts
Then, different promoting procedures were tried to study the influence of the nature of the Co precursor (inorganic, organic) and of the operational conditions on the catalytic activities of supported CoMoS catalysts. Results obtained at 613 K are presented in Table 3. On titania, CoMoS catalysts lead roughly to the same activities whatever the preparation method used or the cobalt source. On silica and γ-alumina, both the preparation method and the cobalt source used have a strong influence on the catalytic activity. The addition of Co(NO)3 on sulfide form of molybdenum enhanced greatly the catalytic activity in the case of silica, while on γ-alumina the effect of the preparation method is less pronounced (Fig. 1). Among these catalytic systems the Co(acac) promoted catalyst supported on γ-Al2O3 gave the highest catalytic activity. Concerning the traditional preparation addition of Co(NO)3 on supported MoOx a synergetic factor defined as Vi(promoted catalyst)/Vi(unpromoted catalyst) of 7 was obtained on γ-Al2O3 (Table 3) while on TiO2 and SiO2 lower synergetic factors were obtained (3 and 2, respectively). However, upon addition of Co promoter, using (Co(NO3)2·6H2O) as cobalt source, on supported molybdenum sulfide, only slightly higher activities were obtained on TiO2, while better synergetic factors were observed on SiO2 and on γ-Al2O3 (Fig. 2 and Table 3) [23,24]. Finally, upon promotion with the organometallic compound cobalt acetylacetonate, impregnated via the refluxing method in methanol, the catalytic activities of supported CoMoS phase on SiO2 and γ-Al2O3 strongly increases and synergetic factors of 9.2 and 17.9 were respectively obtained (Table 3). On TiO2, the promotion with Co(acac)2·nH2O gave no enhancement (similar activities and synergetic factor) compared to the use of cobalt nitrate as Co source (Fig. 2).
Catalytic activities of the Co-promoted molybdenum catalysts.
| Catalysts | Rate constant (V) for thiophene HDS at 613 K (10−8 mol g−1 s−1) | Rate constant (Vi) for thiophene HDS at 613 K (10−4 molecule at Mo−1 s−1) | αa |
| CoMoS–O/TiO2 | 229 | 37 | 3.0 |
| CoMoS–S/TiO2 | 245 | 39 | 3.3 |
| CoMoS–acac/TiO2 | 263 | 42 | 3.4 |
| CoMoS–O/SiO2 | 113 | 9 | 1.9 |
| CoMoS–S/SiO2 | 268 | 22 | 4.6 |
| CoMoS–acac/SiO2 | 530 | 42 | 9.2 |
| CoMoS–O/γ-Al2O3 | 372 | 32 | 7.1 |
| CoMoS–S/γ-Al2O3 | 506 | 37 | 8.3 |
| CoMoS–acac/γ-Al2O3 | 1143 | 79 | 17.9 |
a Synergetic factor: α = Vi(CoMoS/support)/Vi(MoS2/support).

Thiophene HDS activities on supported CoMoS prepared with Co(NO)3 on supported MoOx or MoS2.
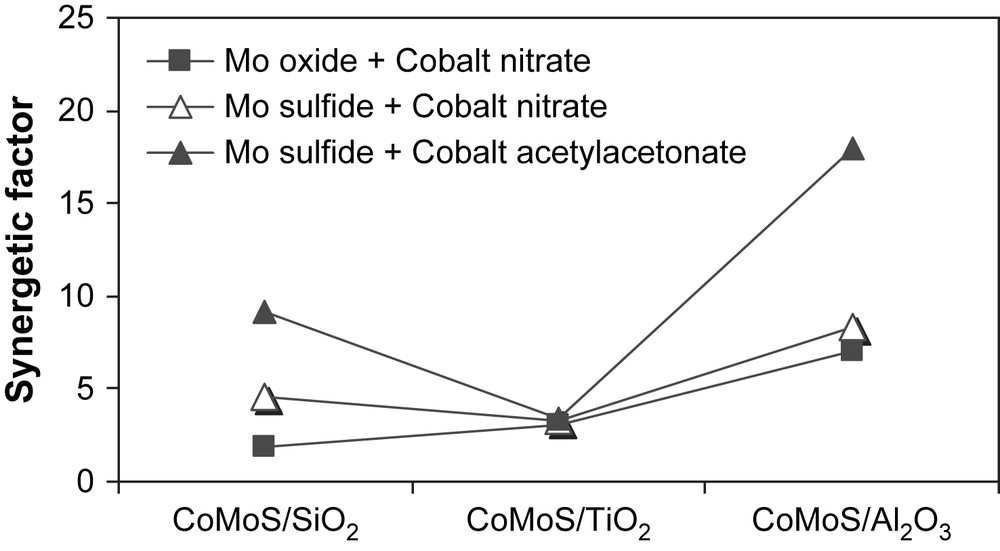
Thiophene HDS synergetic factors obtained on supported CoMoS catalysts compared to supported MoS2.
3.4 Electron microscopy results
The high-resolution electron microscopy analyses carried out on the unpromoted molybdenum sulfide catalysts gave information about the distribution of slab length (L) and the number of fringes (N). Results are presented in Table 4. Fig. 3 shows pictures of typical MoS2 crystallites on MoS2/TiO2 (A), MoS2/SiO2 (B) and MoS2/γ-Al2O3 (C). The distribution of slab's length and number of fringes for supported unpromoted catalysts are indicated in Figs. 4 and 5, respectively. On MoS2/TiO2 catalyst, the distribution of crystallites length was shifted toward the smaller particles (average length = 2.9 nm) as compared with the γ-alumina (average length = 3.7 nm) and silica (average length = 4.5 nm) supported MoS2 catalysts (Fig. 4). Additionally, the MoS2/TiO2 catalyst presents crystallites with a smaller number of fringes (average fringes = 2.1) than γ-Al2O3 (average fringes = 2.8) and SiO2-supported (average fringes = 3.9) MoS2 catalysts (Fig. 5). These results concerning the length and the number of fringes distributions on the different supported molybdenum sulfide are in good agreement with literature data published previously by Vrinat et al. [25].
Average slab length and fringes of supported molybdenum catalysts.
| Catalysts | Average length (nm) | Average fringes |
| MoS2/TiO2 | 2.9 | 2.1 |
| MoS2/SiO2 | 4.5 | 3.9 |
| MoS2/γ-Al2O3 | 3.7 | 2.8 |
| CoMoS/γ-Al2O3 | 3.2 | 2.4 |
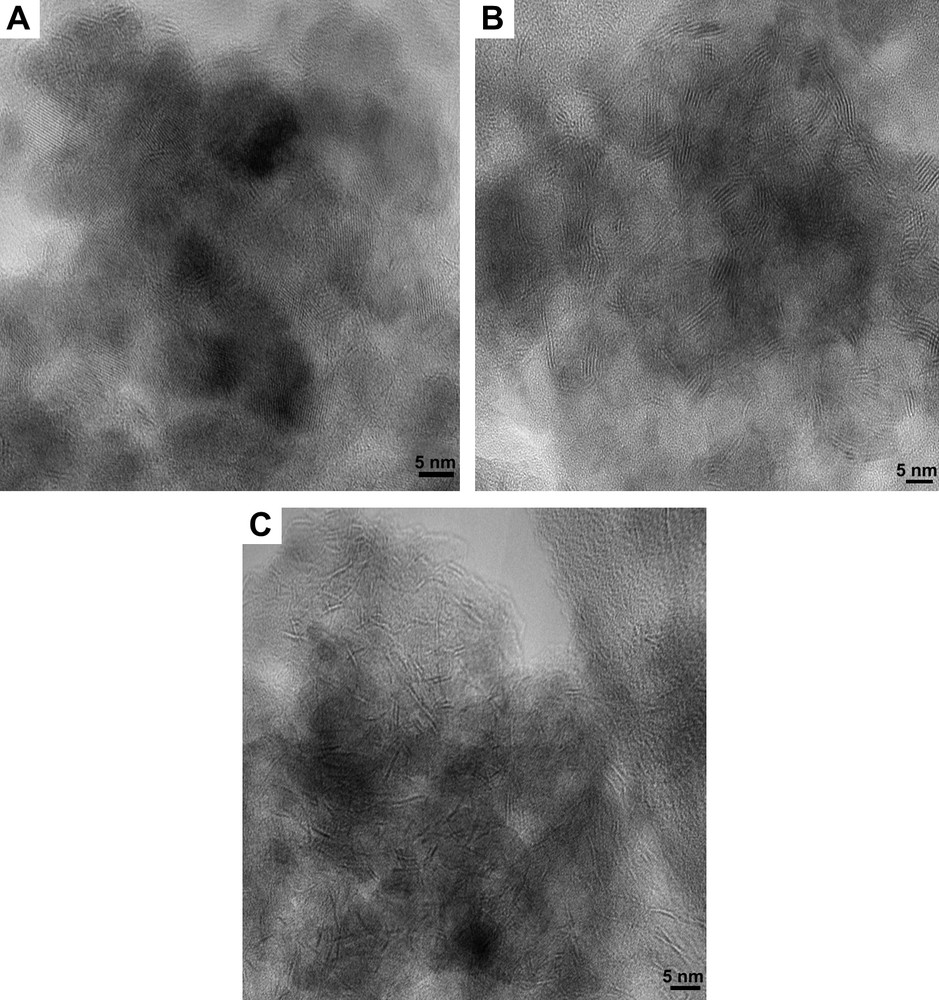
HREM micrograph of: (A) MoS2/TiO2 catalyst. (B) MoS2/SiO2 catalyst. (C) MoS2/γ-Al2O3 catalyst.
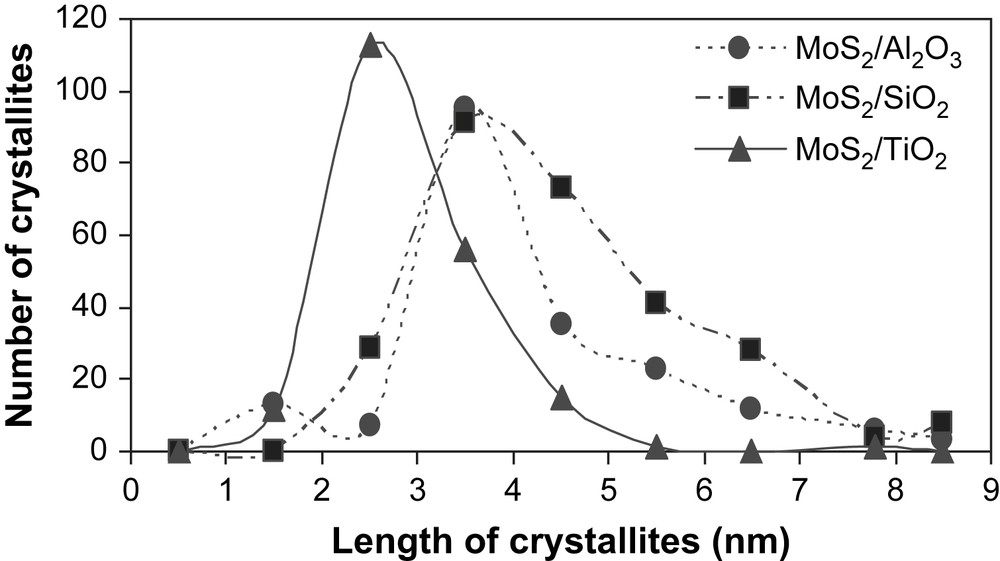
Crystallite length distribution for γ-Al2O3, TiO2 and SiO2-supported unpromoted catalysts.
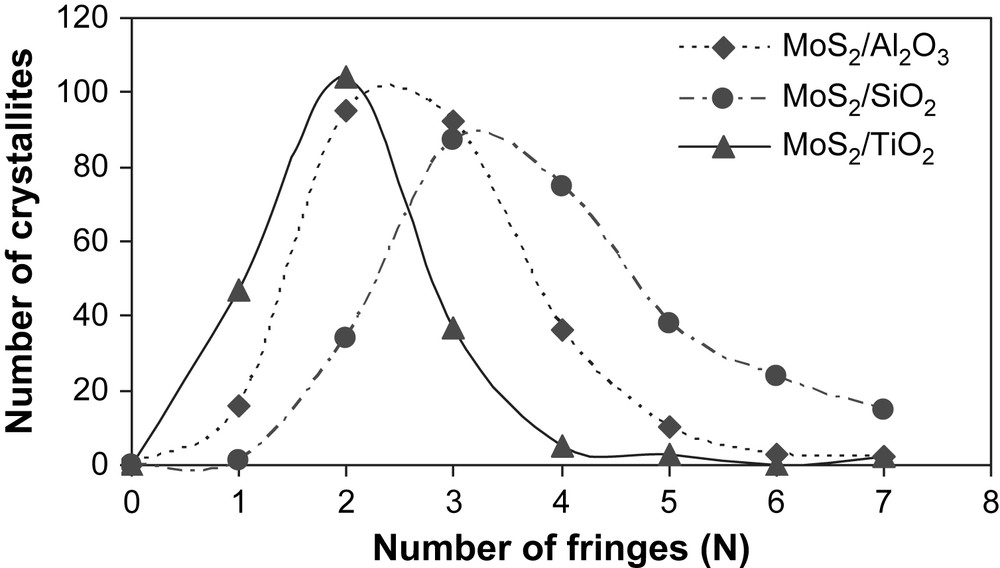
Crystallite fringes distribution for γ-Al2O3, TiO2 and SiO2-supported unpromoted catalysts.
3.5 X-ray photoelectron spectroscopy results
Figs. 6–8 show the decomposition of Mo 3d spectra for MoS2/TiO2, MoS2/SiO2 and MoS2/γ-Al2O3 respectively. All these Mo 3d spectra can be described in terms of Mo6+, Mo5+ and Mo4+ doublets. The Mo4+ state corresponding to MoS2 was obtained as the major Mo specie (80–85%, Table 5) which means that the catalysts are well sulfided. More precisely, the resulted XPS spectra highlight, whatever the support, the presence of essentially one doublet with a Mo 3d5/2 binding energy of 229.0 ± 0.1 eV on SiO2 (80%) and γ-Al2O3 (85%) or 228.6 eV on TiO2 (81%). These values are in good agreement with those expected for the Mo4+ centers in MoS2 [15,26–31]. Moreover, in addition to Mo4+ species there is a small amount of residual molybdenum oxide in a formal 6+ oxidation state (Mo6+) with a Mo 3d5/2 binding energy of 232.5 ± 0.1 eV on SiO2 (5%) and γ-Al2O3 (6%) or 231.6 eV on TiO2 (8%) [26–28]. One additional doublet with a Mo 3d5/2 binding energy of 230.25 ± 0.1 eV on SiO2 (15%) and γ-Al2O3 (9%) or 229.6 eV on TiO2 (11%) was highlighted. This doublet reveals the presence of Mo5+ species and was attributed to the presence of Mo species in an oxysulfide environment [15,26–28]. Finally, it appears from these decompositions that two different sulfur species are present at lower binding energies (226.1 ± 0.3 eV and 227.2 ± 0.5 eV) which are attributed to S2− (MoS2) and S22− species [27], respectively. Similarly, through S 2p core peaks examination, S2− and S22− species have been identified (not given).
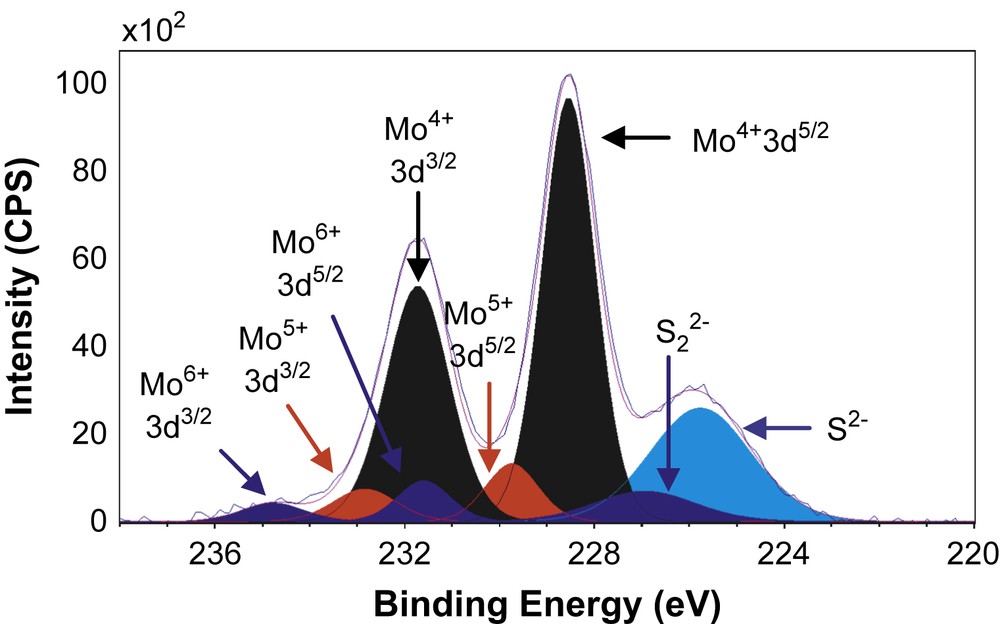
XPS Mo 3d spectrum of MoS2/TiO2.
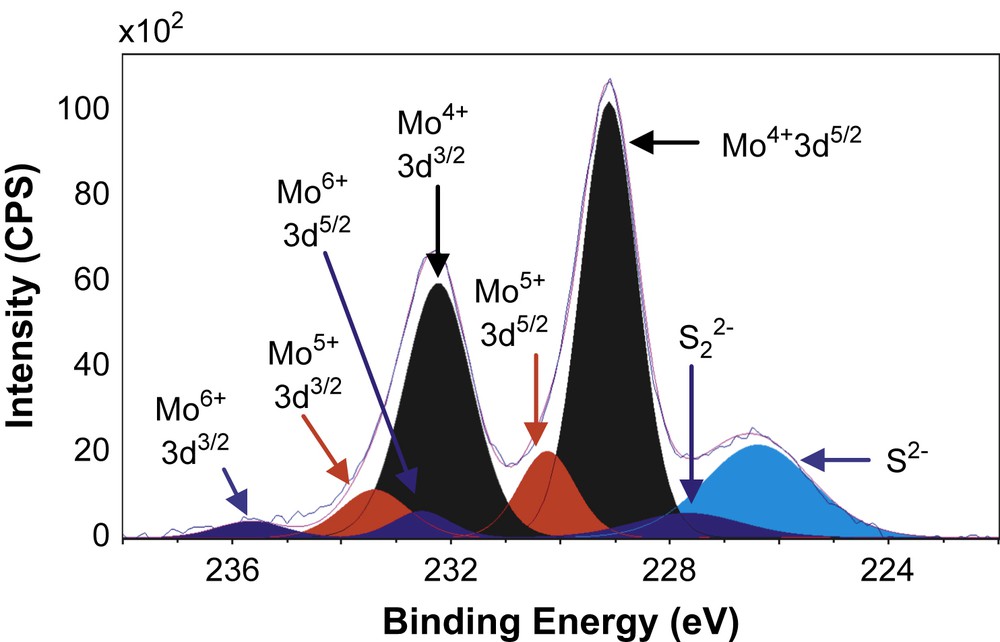
XPS Mo 3d spectrum of MoS2/SiO2.
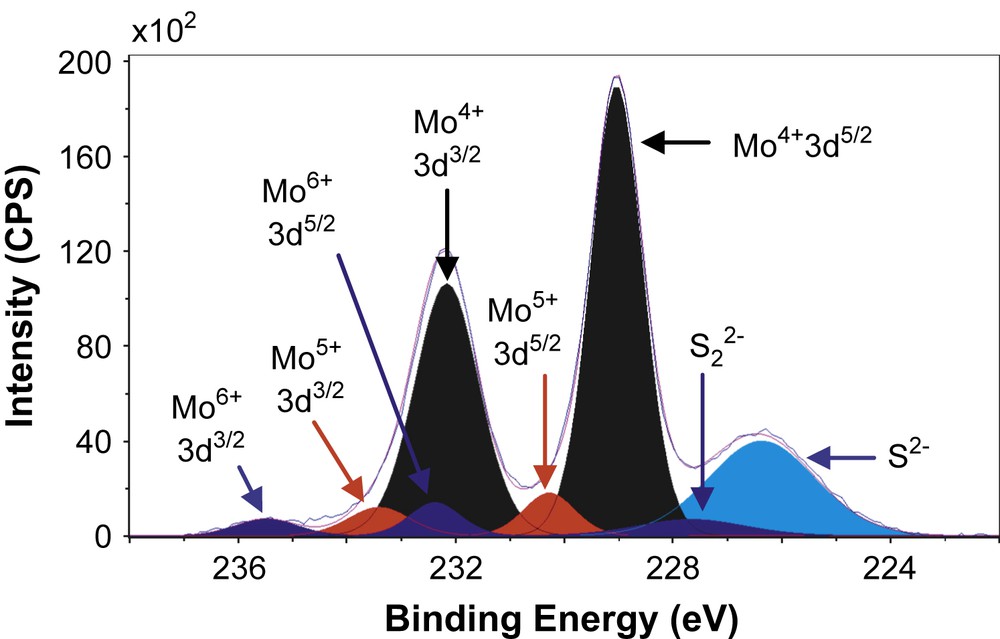
XPS Mo 3d spectrum of MoS2/γ-Al2O3.
XPS results concerning Mo 3d and S 2s spectra.
| Catalysts | Mo4+ | Mo5+ | Mo6+ | S2− | S22− | ||||||
| 3d5/2 | 3d3/2 | % | 3d5/2 | 3d3/2 | % | 3d5/2 | 3d3/2 | % | 2s | 2s | |
| MoS2/TiO2 | 228.5 | 231.6 | 81 | 229.6 | 232.8 | 11 | 231.6 | 234.8 | 8 | 225.8 | 226.7 |
| MoS2/SiO2 | 229.1 | 232.2 | 80 | 230.2 | 233.4 | 15 | 232.6 | 235.7 | 5 | 226.4 | 227.7 |
| MoS2/γ-Al2O3 | 229.1 | 232.2 | 85 | 230.3 | 233.4 | 9 | 232.4 | 235.5 | 6 | 226.4 | 227.7 |
4 Discussion
In the present work, we synthesized a series of unpromoted and promoted catalysts supported on different carriers. Concerning the MoS2 catalysts (Fig. 1), it appeared that better specific and intrinsic activities were obtained with MoS2/TiO2, in a good agreement with the literature [16,19]. In order to explain the better activities obtained on TiO2, the supported unpromoted catalysts were characterized by high-resolution electron microscopy and X-ray photoelectron microscopy. Thus, HREM analyses reveal that on TiO2 the resulted crystallites had smaller number of fringes (average fringes = 2.1) as well as smaller particles (average length = 2.9 nm) by comparison to SiO2 (average fringes = 3.9, average length = 4.5 nm) and γ-Al2O3 (average fringes = 2.8, average length = 3.7 nm) (Table 4). This result indicates out that the higher intrinsic and specific activities observed on TiO2 would be correlated to the lower stacking as well as to the smaller crystallite sizes obtained on this support. Nevertheless, Vrinat et al. [32] showed that for catalysts in which crystallites size vary from 3 to 5 nm and crystallites stacking from 2 to 4 slabs, the ratios Moedge/Mototal or Moedge/Mototal × N (N being the average stacking, Moedge being the presumed catalytically active sites) do not significantly change whatever the catalyst. Therefore, the better catalytic activities observed on TiO2 cannot be only explained by the differences in the crystallites length and slabs stacking.
Similarly, XPS analyses did not allow us to explain the different catalytic activities of unpromoted molybdenum catalysts. More precisely, the different recorded Mo 3d spectra showed that all the catalysts contain similar mixtures of three contributions corresponding to different surface molybdenum species (Mo6+, Mo5+ and Mo4+) and that their molybdenum atoms are mainly in their sulfided form “MoS2” (80–85% of MoS2, Table 5). Furthermore, by changing the support no significant differences in binding energies were observed which suggest that there is no effect of support on the electronic state of surface molybdenum species to explain the better catalytic activities of MoS2/TiO2.
When promotion is performed by impregnation of cobalt nitrate on γ-Al2O3 supported molybdenum oxide, the resulted catalyst presents good synergetic effect. Nevertheless, on TiO2 and SiO2 lower synergetic effects were observed. These results, especially on TiO2, are in good agreement with the observation made previously where it was proposed that a segregation of phase between molybdenum sulfide and the promoter occurs after sulfidation leading to a low synergetic effect. This assumption is still unclear and strongly discussed in the literature [16,17,19].
However, by adding the cobalt nitrate promoter on the molybdenum sulfide phase, better catalysts were obtained on γ-Al2O3 and SiO2. The microscopy analyses of the CoMoS/γ-Al2O3 have allowed obtaining similar values for slabs' lengths and stacking than for MoS2/γ-Al2O3, thus the difference in catalytic activity could not be only explained by the morphology. On the other hand, no further enhancement on TiO2 was observed compared to the previous method. The better catalytic activities (Fig. 1) obtained when the Co promoter is added on the molybdenum sulfide phase could be explained, according to the literature, by a more favorable formation of the mixed CoMoS phase. This is probably due to the tendency of the group VII metal, like cobalt, to be sulfided before molybdenum giving isolated cobalt sulfide crystallites “Co9S8” not active in HDS reaction, in a classical preparation procedure [33].
Recently, it was assumed by Bezverkhyy et al. that starting from MoS2 and by using Co(NO3)2·6H2O a reaction occurs between molybdenum sulfide and the nitrate (NO3−) anions, leading to a modification of the MoS2 phase structure and consequently influencing all the impregnation procedure [33]. So, in order to avoid the modification of the presulfided molybdenum phase, Co(acac)2·nH2O precursor was chosen to introduce the promoter metal; this way proved to enhance the promotion of the supported MoS2 phase [23]. Thus, in total accordance with these assumptions, it may be observed that except for TiO2 support, the use of cobalt acetylacetonate gave higher catalytic activities in thiophene hydrodesulfurization compared with the supported CoMoS phases prepared via the conventional cobalt nitrate route (Fig. 2).
These results strongly suggest that the promoting method has a considerable effect on catalytic activity for the catalysts prepared on SiO2 and γ-Al2O3. On γ-Al2O3, the promoting effect can easily be increased by a factor of 3. Concerning TiO2, the same synergetic factor was obtained whatever the promoting method used. A parallelism could be established between this particular MoS2/TiO2 crystallite behavior and recent DFT calculations realized by Raybaud et al. [34,35]. According to these authors, MoS2 crystallites present two types of sites: metallic edges (Mo-edge) and sulfur edges (S-edge) sites which exhibit different chemical interactions with the support. Therefore, the higher activity observed for MoS2/TiO2 as compared to MoS2/γ-Al2O3 would be correlated to a higher Mo-edge/S-edge ratio on TiO2 than on γ-Al2O3 and a strong ligand effect of TiO2 anatase which stabilize sulfur deficient particles [35]. Furthermore, DFT calculations [36] realized on Co-promoted catalysts showed that cobalt have a preferential affinity for the S-edge sites which means that Mo-edge sites on TiO2 have a very weak interaction with cobalt, thus they are unfavorable for cobalt decoration. This could explain the low synergetic effect observed for the catalysts CoMoS/TiO2 prepared in this study compared to CoMoS/γ-Al2O3.
5 Conclusion
It has been shown in this study that adding directly the cobalt promoter on the molybdenum sulfide phase, lead to higher synergetic effects for SiO2 and γ-Al2O3 supports in thiophene HDS. Additionally, compared to catalysts prepared by a classical impregnation method, better hydrodesulfurization catalysts were obtained on γ-Al2O3 and SiO2 when an organic promoter such as cobalt acetylacetonate was used. This result confirms the possibility to control the formation of the active phase: better promotion of MoS2 and less parasitic phases. On TiO2, changing the cobalt precursor and the promoting method has no significant effect on catalytic activities. This TiO2 behavior is probably due to the MoS2 slab orientation where primarily metallic Mo edges sites are exposed. With such sites and when cobalt is used as promoter, the cobalt decoration is unfavorable, leading to a less-active hydrodesulfurization catalysts. Further characterizations are underway to obtain more structural information on these systems including for instance, infrared and X-ray photoelectron spectroscopies of promoted catalysts.
Acknowledgement
The authors would like to thank the Centre national de la recherche scientifique and the Agence nationale de la recherche for financial supports (ANR. INNOVCAT. n° 0136).