

1 Introduction
The naturally occurring Substance P (SP) is an undecapeptide belonging to the tachykinine family of peptides, with very important biological activities [1,2]. It was established that the release of SP is related to the transmission of pain and the induction of neurogenic inflammatory response [2–4]. SP evokes the release of such neurotransmitters as acetylholine, histamine and GABA [5]. The low degree of receptor selectivity combined with the metabolic instability of peptides limit their use as pharmacological tools [1]. This stimulated the search for potent nonpeptide antagonists of the human neurokinin-1 (NK1) receptor to which SP preferentially binds [6]. It has recently been reported that 3-amino- or 3-hydroxypiperidine derivatives such as CP-99994 and L-733060 respectively, exhibit excellent affinity and selectivity with human NK1 receptor [1,2,6–8]. Piperidinones are also useful advanced intermediates in the preparation of piperidines [9] and scaffolds in the synthesis of β-turn peptide mimetics [10]. Incorporation of an α-amino acid residue into the piperidinone ring gave rise to the so-called constraint pseudopeptides. Some of them have been shown to possess ACE inhibitory activity [11,12]. Our interest was focused on substituted piperidinones of type I and II (Fig. 1), because of their structural similarity with the already known SP antagonists.
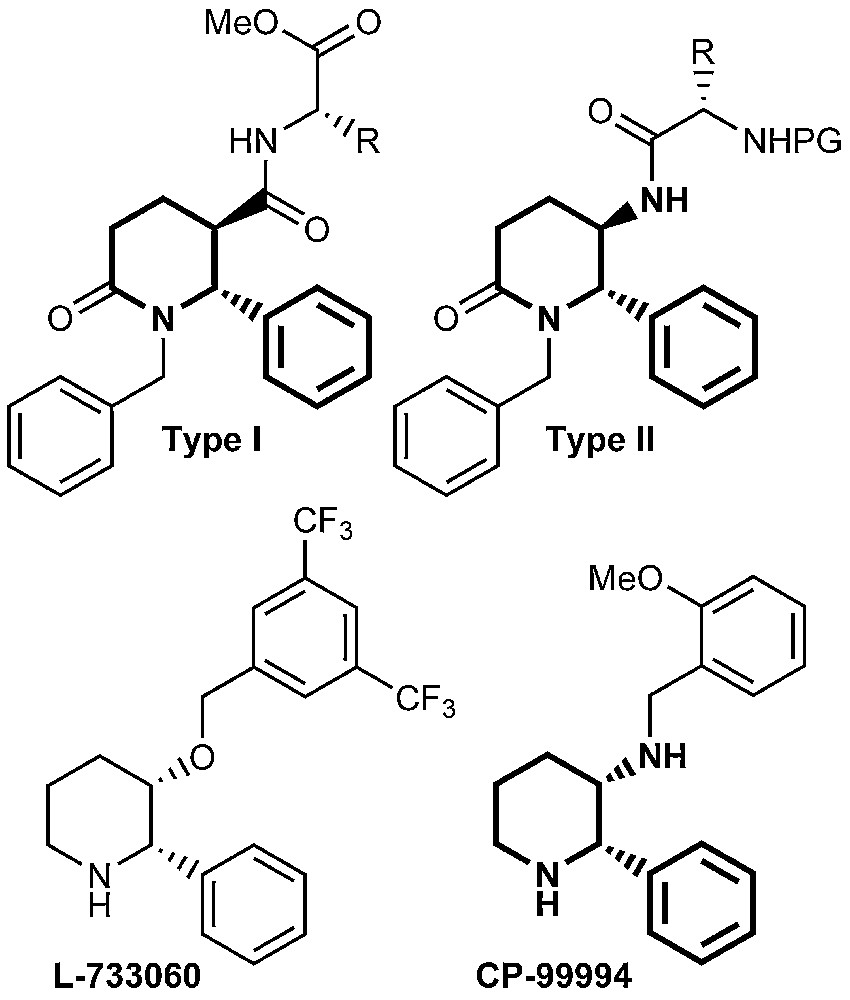
Substituted piperidinones of types I and II.
Although the disubstituted piperidine framework has shown various pharmaceutical activities, the pattern of disubstituted piperidines of types I and II is less explored [5].
The purpose of the present investigation is to incorporate a moiety of an amino acid via a peptide bond to the piperidinone ring leading to 3-carboxamides of type I and 5-(acylated amino)piperidinones of type II. This structural modification would allow the investigation of the binding ability of the newly synthesized compounds with the human NK1 receptor. Glycine and L-α-phenylalanine were used, because they are components of the structure of SP. Protected L-tryptophan was selected as well, because esters of N-acetyl-L-tryptophan are shown to possess high binding affinity towards human NK1 receptor [1,13,14].
2 Results and discussion
The first step in the preparation of compounds of type I and II is the formation of the piperidinone ring. The reaction of glutaric anhydride with N-arylidene-N-alkylamines provides a straightforward path to the synthesis of the piperidinone ring, affording many racemic trans-1-alkyl-2-aryl-6-oxopiperidine-3-carboxylic acids in one step [15–18]. The synthesis of (±)-trans-1-benzyl-6-oxo-2-phenylpiperidine-3-carboxylic acid (1) and its acid chloride 2 was previously described [15,17,19]. Acid 1 is a suitable starting compound for carboxylic group transformations into either 3-carboxamido compounds or into 5-amino derivatives. As was already mentioned, acid 1 is obtained as a racemic mixture of enantiomers and only one of them will be shown throughout the text for clarity. Two reaction pathways were explored, and they are depicted on Scheme 1. The reactions shown do not affect the chiral centres of the heterocycle and thus the trans relative configuration of the products follows directly from the configuration of the starting acid 1.
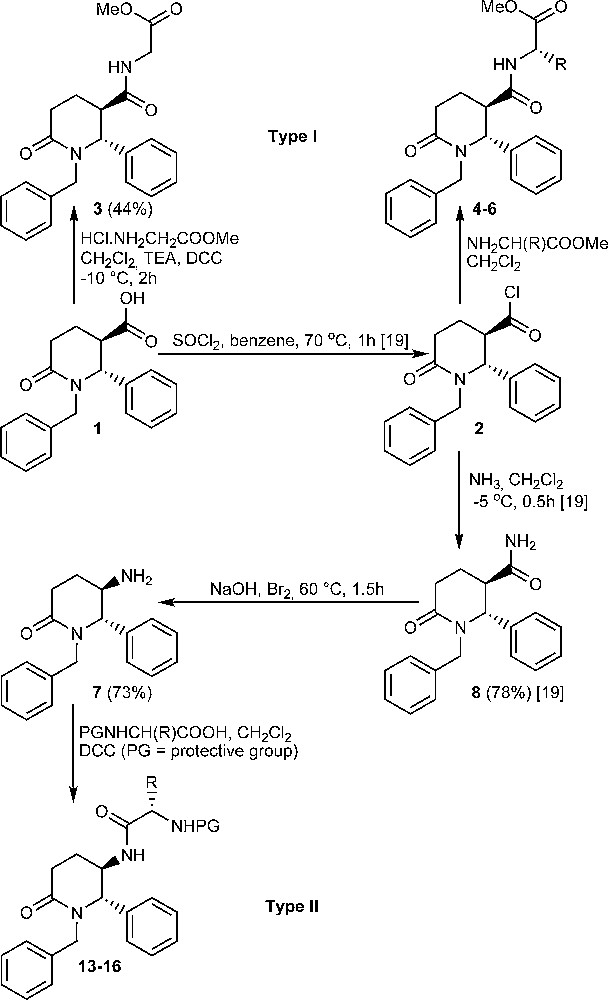
Synthesis of compounds of type I and II (only one enantiomer is shown).
The synthesis of compounds of type I involves a peptide bond construction between acid 1 as an acylation agent and methyl esters of glycine, L-α-alanine, L-α-phenylalanine and L-tryptophan. Since the acid 1 was shown to give readily the acid chloride 2 [19], we attempted first N-acylation of the selected L-α-amino acid esters with the aid of 2. Methyl esters of L-α-tryptophan and L-α-phenylalanine, were first converted from hydrochlorides to free bases and then treated with the acid chloride 2. Methyl L-α-alaninate was used as a hydrochloride and its acylation by means of 2 was performed in the presence of excess of triethylamine (TEA) as a base to bind hydrogen chloride. Methyl glycinate hydrochloride was treated with the acid 1 in presence of dicyclohexylcarbodiimide (DCC) and TEA. Thus compounds 3–6 were obtained in very good yields after purification by means of column chromatography followed by a recrystallization. Compounds 4–6 are shown in Table 1. Their structures were confirmed by means of elemental analysis, IR and 1H NMR spectra.
Compounds 4–6 and their yields.
| Compound number | R | Yield, % |
| 4 | -CH3 | 70 |
| 5 | -CH2Ph | 52 |
| 6 | -CH2Ind | 74 |
Compounds 4–6 derived from the acid 1 are 1:1 mixtures of α-S, (±)-trans diastereomers as evidenced by their 1H NMR spectra. We did not manage to separate the two diastereomers of compounds 4–6 by means of column chromatography. After fractional recrystallization, compound 4 was separated into two portions, which have close melting points. The 1H NMR spectra of the two portions revealed that the first portion consisted of diastereomers in 20:1 ratio and for the second one the respective ratio was 1:4, determined by the integral for the doublet signals of methyl groups. This allowed us to distinguish the signals of each diastereomer. It was established by this experiment that signals for the same protons in the different diastereomers are not shifted in one and the same direction. With this in mind, in the experimental section we noted the signals for the same protons which have higher chemical shift with “*” symbol. Relative trans configuration of the substituents at the piperidinone ring of peptide derivatives 3–6 was assigned on the basis of the value of coupling constants 3J2,3, which is in the interval of 6.6–8.3 Hz.
We investigated the formation of (±)-trans-5-amino-1-benzyl-6-phenylpiperidin-2-one (7) from the acid 1 using the Hofmann rearrangement protocol [20] from amide 8 [19]. We investigated several types of reaction conditions: the oxidative rearrangement by means of lead tetraacetate [21], the application of diacetoxyiodobenzene [22,23] and the classical protocol, by treatment with bromine in aqueous sodium hydroxide solution [20]. The latter reagent proved to work well in our hands especially when 1–2 mmol of the starting amide 8 were used. When more than 2 mmol of 8 were employed, the reaction rate was slower and the yield of the amine 7 was greatly reduced. The target amine 7, which was obtained as a single diastereomer, is an uncrystallizable oil. However, the purification of 7 by column chromatography was difficult because of its great polarity so in the subsequent acylation reactions it was used without further purification. Amine 7 was characterized by elemental analysis as a crystalline hydrobromide salt. 1H NMR spectrum of amine 7 was taken and compared to 1H NMR data of trans-3-substituted-1-benzyl-2-phenylpiperidin-6-ones investigated previously by us [19]. Amine 7 is characterized by a doublet signal at 4.54 ppm with 3J5,6 = 3.8 Hz for H-6 in its 1H NMR spectrum. Similar values of 3J for disubstituted piperidinones have been previously observed and they are attributed to an equiliblium between two conformations in solution - one with axial-pseudoaxial protons, and the other–with equatorial-pseudoequatorial protons at the chiral centres of the ring [18,19]. This equilibrium appears to be more shifted to the latter conformation. On this ground we attributed trans relative configuration of the substituents at the chiral C-5 and C-6 carbon atoms of the piperidinone ring of 7. Such a conclusion is in agreement with already established retention of the configuration of the migrating carbon atom during the Hofmann rearrangement [20].
As additional proofs for the structure of 7, methylcarbamate 9 and acetamide 10 were also prepared by acylation of 7 with methyl chloroformate and acetic anhydride, resp. (Scheme 2) Derivatives 9 and 10 are crystalline solids, which were characterized by means of mps, elemental analysis and 1H NMR spectra. The trans relative configuration of 9 and 10 was assigned on the base of the singlet signals of H-2 protons in 1H NMR spectra, which can be explained with a preferred conformation with equatorial-pseudoequatorial protons at the chiral centres. This conclusion seems to be relevant, having in mind that compounds 9 and 10 are directly derived from the amine 7. We attempted the preparation of carbamate 9 by a Curtius rearrangement as well [24]. Thus, hydrazide 11 was prepared and treated with nitrous acid at 0 °C (Scheme 3).
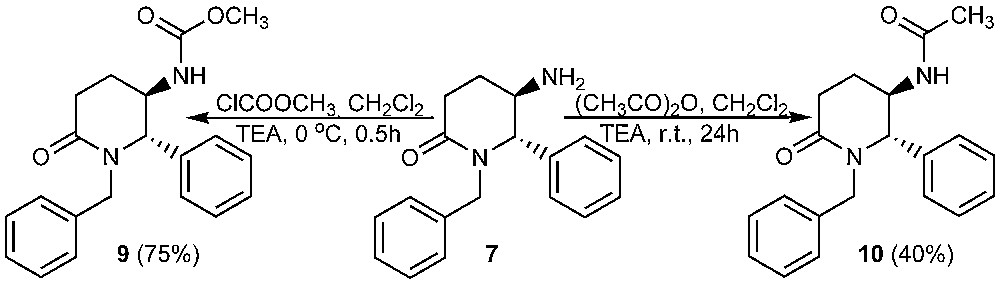
Synthesis of compounds 9 and 10 (only one enantiomer is shown).

Synthesis of compound 9 via Curtius rearrangement (only one enantiomer is shown).
The formation of acyl azide 12 was confirmed by its IR spectrum which exhibited an intensive band for the N3 group at 2130 cm−1. Azide 12 was not stable and without further purification was refluxed in dry methanol. The yield of methyl carbamate 9 is comparable to that obtained via the Hofmann rearrangement. The latter is however more straightforward and on this ground Curtius rearrangement of acyl azide 12 was no longer explored for the purposes of this investigation.
Further amine 7 was reacted with N-protected L-α-amino acids – Cbz-Glycine, N-trifluroacetyl L-α-phenylalanine, Boc-L-tryptophane and Boc-L-α-alanine using the carbodiimide method (Scheme 1) [5,25]. In this way compounds 13–16 were obtained in good yields after column chromatography purification (Table 2).
Compounds 13–16 and their yields.
| Compound number | R | Yield, % |
| 13 | -H | 42 |
| 14 | -CH2Ph | 31 |
| 15 | -CH2Ind | 50 |
| 16 | -CH3 | 61 |
As in the case of compounds 4–6, compounds 14–16 were 1:1 mixtures of α-S, (±)-trans-diastereomers, according to their 1H NMR spectra. In spite of our efforts, we could not separate the diastereomeric couples of compounds 14–16 by means of column chromatography. This made difficult the description of 1H NMR spectral data in the Experimental part, because the signals for the same protons in the different diastereomers are not shifted in one and the same direction. For this reason in the experimental part the signals for the same protons with higher chemical shift are noted with “*” symbol, similarly to compounds 4–6. In the 1H NMR spectra of compounds 13–15 the signal for Н-2 appears as a doublet with 3J2,3 in the interval 3.1–3.8 Hz, while for compound 16 the signal for the same proton was singlet. On this base we assigned trans relative configuration of substituents attached to chiral C-2 and C-3 atoms of the piperidinone ring of compounds 13–16.
Biological screening of newly synthesized compounds is now in course.
3 Experimental
Melting points (mps) were taken on a “Boetius” PHMK 05 micro hot-stage apparatus and are not corrected. IR spectra were recorded on a Specord 75 instrument. 1H NMR spectra (250.13 MHz) were obtained on a Bruker Avance DRX-250 spectrometer. The chemical shifts are given in ppm (δ) relative to tetramethylsilane as internal standard and coupling constants (J) are given in Hz. The microanalyses were performed on VarioEL III CHNS/O, Elementar Analysensysteme GmbH at the Faculty of Chemistry, University of Sofia. Thin layer chromatography (tlc) was performed on Merck 1.05554 silica gel 60F254 aluminum plates. Column chromatography purifications were carried out using Riedel-de Haën silica gel S (0.063–0.100 mm).
Compounds 1, 2 and 8 are prepared according to the procedures given in reference [19].
Methyl [((±)-trans-1-benzyl-6-oxo-2-phenylpiperidine-3-carbonyl)amino]acetate (3). To a solution of acid 1 [19] (0.309 g, 1 mmol) in dry dichloromethane (5 mL), triethylamine (0.14 mL, 1 mmol) and methyl 2-aminoacetate hydrochloride (0.126 g, 1 mmol) were added. The reaction mixture was cooled (-10 °C) and DCC (0.275 g, 1.3 mmol) was added in portions. The mixture was stirred for 2 h at -10 °C and then 12 h at room temperature. The resulting precipitate of 1,3-dicyclohexylurea was filtered and discarded. The filtrate was evaporated under reduced pressure and the resulting oil was dissolved in ethyl acetate (30 mL). The solution was successively washed with 10% HCl, water, 10% Na2CO3 and brine. Organic layer was dried (Na2SO4) and the solvent was removed in vacuo. Recrystallization from light petroleum/ethyl acetate yielded 0.167 g (44%), mp 130–131 °C; IR (Nujol): 1610, 1680 and 1750 (C = O), 3280 (NH) cm−1; 1H NMR (CDCl3): δ 1.91–2.13 (m, 2H, H-4), 2.50–2.82 (m, 3H, H-3, 2xH-5), 3.35 (d, 1H, CH2Ph, J = 14.7 Hz), 3.69 (dd, 1H, CH2NH, J = 5.1, 18.2 Hz), 3.69 (s, 3H, OCH3), 3.84 (dd, 1H, CH2NH, J = 5.6, 18.2 Hz), 4.69 (d, 1H, H-2, J = 6.6 Hz), 5.54 (d, 1H, CH2Ph, J = 14.7 Hz), 5.65 (t, 1H, NHCO, J = 5.0 Hz), 7.07–7.42 (m, 10H, Ph). Anal. Calcd. for C22H24N2O4: C, 69.46; H, 6.36; N, 7.36; Found: С, 69.70; Н, 6.37; N, 7.31.
Methyl (2S)-2-[((±)-trans-1-benzyl-6-oxo-2-phenylpiperidine-3-carbonyl)amino]-propanoate (4). A solution of acid chloride 2 [19] (1.5 mmol) in dry dichloromethane (3 mL) was cooled (0 °C) and triethylamine (0.41 mL, 3 mmol) was added to it. Solution of L-α-alanine methyl ester hydrochloride (0.206 g, 1.5 mmol) in dry dichloromethane (3 mL) was added and the reaction mixture was stirred for 0.5 h at room temperature. The solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (50 mL). Organic solution was washed with water (3х20 mL) and dried (Na2SO4). Recrystallization from ethyl acetate afforded 0.189 g (32%) of 20:1 diastereomeric mixture of 4, mp 197–199 °C. Concentration of the filtrate yielded 0.228 g (38%) of 1:4 diastereomeric mixture of 4, mp 199–201 °C. The total yield was 0.417 g (70%) of 4 as white crystals. IR (Nujol): 1635, 1675 and 1735 (С = О), 3425 (NH) cm−1; 1H NMR (CDCl3): δ 1.02 (d, 3H, CHCH3, J = 7.2 Hz), 1.19 (d, 3H, CHCH3*, J = 7.2 Hz), 1.91–2.18 (m, 4H, 2xH-4, 2xH-4*), 2.48–2.83 (m, 6H, H-3, H-3*, 2xH-5, 2xH-5*), 3.37 (d, 1H, CH2Ph, J = 14.7 Hz), 3.39 (d, 1H, CH2Ph*, J = 14.7 Hz), 3.66 (s, 3H, OCH3), 3.69 (s, 3H, OCH3*), 4.40 (dq, 2H, CHCH3, CHCH3*, J = 7.2 Hz), 4.64 (d, 1H, H-2, J = 8.3 Hz), 4.71 (d, 1H, H-2*, J = 6.3 Hz), 5.48 (d, 1H, CH2Ph, J = 14.7 Hz), 5.53 (d, 1H, CH2Ph*, J = 14.7 Hz), 5.61 (d, 1H, NHCO, J = 7.9 Hz), 5.53 (d, 1H, NHCO*, J = 7.2 Hz), 7.03–7.42 (m, 20H, Ph, Ph*). Anal. Calcd for C23H26N2O4: C, 70.03; H, 6.64; N, 7.10; Found: С, 70.67; Н, 6.65; N, 7.40.
General procedure for the synthesis of (±)-trans-1-benzyl-6-oxo-2-phenylpiperidine-3-carboxamides 5 and 6. Hydrochloride of the methyl ester of the corresponding L-α-amino acid (3 mmol) is transformed into a base and extracted with dichloromethane. The organic solution was dried (Na2SO4). The solvent was removed and the residue was dissolved in dry dichloromethane (2 mL). This solution was added dropwise to a cooled (0 °C) solution of acid chloride 2 [19] (1 mmol) in dry dichloromethane (3 mL). Reaction mixture was stirred for 0.5 h at room temperature and solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (50 mL) and the organic solution was washed with water (3х20 mL). Organic layer was dried (Na2SO4). Crude products were purified by means of column chromatography and/or recrystallization from ethyl acetate.
Methyl (2S)-2-(((±)-trans-1-benzyl-6-oxo-2-phenylpiperidine-3-carbonyl)amino)-3-phenylpropanoate (5). Synthesized from acid chloride 2 and L-α-phenylalanine methyl ester. Recrystallization from ethyl acetate yielded 0.245 g (52%) of 5 as white crystals, mp 148–150 °C; IR (Nujol): 1640, 1670 and 1700 (С = О), 3380 (NH) cm−1; 1H NMR (CDCl3): δ 1.79–2.12 (m, 4H, 2xH-4, 2xH-4*), 2.45–2.76 (m, 7H, H-3, H-3*, 2xH-5, 2xH-5*, CHCH2Ph), 2.82 (dd, 1H, CHCH2Ph*, J = 6.8, 13.9 Hz), 2.91 (dd, 1H, CHCH2Ph, J = 4.9, 13.8 Hz), 3.09 (dd, 1H, CHCH2Ph*, J = 5.6, 13.9 Hz), 3.36 (d, 1H, CH2Ph, J = 14.8 Hz), 3.37 (d, 1H, CH2Ph*, J = 14.8 Hz), 3.65 (s, 3H, OCH3), 3.66 (s, 3H, OCH3*), 4.62–4.81 (m, 4H, H-2, H-2*, CHNH, CHNH*), 5.45 (d, 1H, CH2Ph, J = 14.8 Hz), 5.48 (d, 1H, CH2Ph*, J = 14.8 Hz), 5.58–5.73 (m, 2H, NHCO, NHCO*), 6.39–6.50 (m, 2H, Ph, Ph*), 6.88–7.43 (m, 28H, Ph, Ph*). Anal. Calcd. for C29H30N2O4: C, 74.02; H, 6.43; N, 5.95; Found: С, 74.60; Н, 6.41; N, 5.92.
Methyl (2S)-2-(((±)-trans-1-benzyl-6-oxo-2-phenylpiperidine-3-carbonyl)amino)-3-(1H-indol-3-yl)propanoate (6). Synthesized from acid chloride 2 and L-tryptophan methyl ester. Column chromatography with light petroleum:ethyl acetate (1:2, v/v) and further recrystallization from ethyl acetate afforded 0.378 g (74%) of 6 as white crystals, mp 112–114 °C; IR (Nujol): 1620, 1650 and 1750 (С = О), 3190 and 3410 (NH) cm−1; 1H NMR (DMSO-d6): δ 1.55–1.91 (m, 4H, 2xH-4, 2xH-4*), 2.26–2.57 (m, 4H, 2xH-5, 2xH-5*), 2.66–3.01 (m, 5H, H-3, H-3*, 2xCH2Ind, CH2Ind*), 3.10 (dd, 1H, CH2Ind*, J = 5.4, 14.5 Hz), 3.31 (d, 1H, CH2Ph, J = 15.4 Hz), 3.33 (d, 1H, CH2Ph*, J = 15.4 Hz), 3.49 (s, 3H, OCH3), 3.51 (s, 3H, OCH3*), 4.34–4.49 (m, 2H, CHNH, CHNH*), 4.62 (d, 1H, H-2, J = 6.6 Hz), 4.65 (d, 1H, H-2*, J = 6.6 Hz), 5.15 (d, 1H, CH2Ph, J = 15.4 Hz), 5.17 (d, 1H, CH2Ph*, J = 15.4 Hz), 6.70–7.47 (m, 30H, Ph, Ph*), 8.32 (d, 1H, NHCO, J = 7.5 Hz), 8.35 (d, 1H, NHCO*, J = 8.0 Hz), 10.77 (d, 1H, indolic NH, J = 1.7 Hz), 10.85 (d, 1H, indolic NH*, J = 1.8 Hz). Anal. Calcd. for C31H31N3O4: C, 73.06; H, 6.13; N, 8.25; Found: С, 73.26; Н, 6.10; N, 8.08.
Synthesis of (±)-trans-5-amino-1-benzyl-6-phenylpiperidin-2-one (7). To a cooled (-5 °C) solution of NaOH (0.480 g, 12 mmol) in water (6 mL), bromine (0.12 mL, 2.3 mmol) was added. The mixture was stirred for 0.5 h and (±)-trans-1-benzyl-6-oxo-2-phenylpiperidine-3-carboxamide 8 [19] (0.616 g, 2 mmol) was added. The reaction mixture was stirred at 60 °C for 1.5 h until completion of the reaction (tlc). After extraction with ethyl acetate combined organic layers were washed with 1:1 HCl (25 mL). The water layer was then alkalized with 10% NaOH and extracted with ethyl acetate. Organic phase was dried (Na2SO4) and solvent was evaporated under vacuo. The oily product 0.410 g (73%) was used for further transformations without additional purification. IR (Nujol): 1620 (C = O), 3360 and 3480 (NH2) cm−1.
Synthesis of (±)-trans-5-amino-1-benzyl-6-phenylpiperidin-2-one hydrobromide. Amine 7 (0.450 g, 1.6 mmol) was dissolved in dry methanol (5 mL) and 48% hydrobromic acid was added until pH = 3. Dry diethyl ether was added (1 mL) and crystallized solid was collected by filtration. Yield 0.448 g (78%), mp 175–178 °C; IR (Nujol): 1595 (NH), 1620 (C = O), 2450–3250 (NH3+) cm−1; 1H NMR (DMSO-d6): δ 1.70–1.86 (m, 1H, H-4), 1.87–2.04 (m, 1H, H-4), 2.55–2.71 (m, 2H, H-3), 3.42 (d, 1H, CH2Ph, J = 15.1 Hz), 3.49–3.60 (m, 1H, H-5), 4.54 (d, 1H, H-6, J = 3.8 Hz), 5.15 (d, 1H, CH2Ph, J = 15.1 Hz), 7.06–7.48 (m, 10H, Ph), 8.06 (br. s, 3H, NH3+). Anal. Calcd. for C18H20N2O.HBr: C, 59.84; H, 5.86; N, 7.75; Found: С, 60.28; Н, 5.84; N, 7.84.
Methyl (±)-trans-1-benzyl-6-oxo-2-phenylpiperidin-3-ylcarbamate (9). To a cooled (0 °C) solution of amine 7 (0.311 g, 1.1 mmol) in dichloromethane (5 mL), triethylamine (0.21 mL, 1.5 mmol) and methyl chloroformate (0.1 mL, 1.3 mmol) were added. Reaction mixture was stirred for 0.5 h at room temperature until completion of the reaction (tlc). The mixture was diluted with ethyl acetate (40 mL) and washed with water, 10% Na2CO3 and water till pH = 7. Organic phase was dried (Na2SO4) and the solvent was evaporated under reduced pressure. Column chromatography with light petroleum:2-propanol (20:1, v/v) yielded 0.277 g (75%) of 9 as white amorphous product, mp 60–65 °C; IR (CHCl3): 1630, 1640 and 1715 (С = О), 3435 (NH) cm−1; 1H NMR (CDCl3): δ 1.63–1.77 (m, 1H, H-4), 1.85–2.02 (m, 1H, H-4), 2.49–2.74 (m, 2H, H-5), 3.35 (d, 1H, CH2Ph, J = 14.6 Hz), 3.55 (s, 3H, OCH3), 3.83–3.99 (m, 1H, H-3), 4.57 (s, 1H, H-2), 4.87–5.03 (m, 1H, NHCO), 5.67 (d, 1H, CH2Ph, J = 14.6 Hz), 7.12–7.45 (m, 10H, Ph). Anal. Calcd. for C20H22N2O3: C, 70.99; H, 6.55; Found: C, 70.57; H, 6.55.
N-((±)-Trans-1-benzyl-6-oxo-2-phenylpiperidin-3-yl)acetamide (10). To a solution of amine 7 (0.258 g, 1 mmol) in dry dichloromethane (21 mL), triethylamine (0.17 mL, 1.2 mmol) and acetic anhydride (0.1 mL, 1 mmol) were added. Reaction mixture was stirred for 24 h at room temperature, diluted with ethyl acetate (50 mL) and washed with water (3х15 mL). Organic phase was dried (Na2SO4) and the solvent was evaporated. Recrystallization from methanol/water yielded 0.120 g (40%) of 10 as white crystals, mp 186–188 °C; IR (Nujol): 1640 (C = O), 3330 (NH) cm−1; 1H NMR (CDCl3): δ 1.56–1.70 (m, 1H, H-4), 1.81 (s, 3H, CH3CO), 1.82–1.95 (m, 1H, H-4), 2.41–2.65 (m, 2H, H-5), 3.35 (d, 1H, CH2Ph, J = 14.4 Hz), 4.14–4.24 (m, 1H, H-3), 4.62 (s, 1H, H-2), 5.63 (d, 1H, CH2Ph, J = 14.4 Hz), 5.99 (d, 1H, NHCO, J = 7.2 Hz), 7.18–7.46 (m, 10H, Ph). Anal. Calcd. for C20H22N2O2: C, 74.51; H, 6.88; Found: С, 74.40; Н, 6.97.
(±)-Trans-1-benzyl-6-oxo-2-phenylpiperidin-3-carbohydrazide (11). A solution of methyl (±)-trans-1-benzyl-6-oxo-2-phenylpiperidin-3-carboxylate [19] (0.161 g, 0.5 mmol) and hydrazine hydrate (0.25 g, 5 mmol) in dry methanol (1 mL) was refluxed for 5 h. The solvent was evaporated and the residue was dissolved in dichloromethane and dried (Na2SO4). The solvent was removed and the residue was recrystallized from ethyl acetate, which yielded 0.137 g (85%) of 11 as white crystals, mp 143–144 °C; IR (Nujol): 1640 and 1660 (С = О), 3200 and 3330 (NH) cm−1; 1H NMR (CDCl3): δ 1.89–2.20 (m, 2H, H-4); 2.46–2.86 (m, 3H, H-3, 2xH-5); 3.40 (d, 1H, CH2Ph, J = 14.7 Hz); 3.68 (broad s, 2H, NH2); 4.66 (d, 1H, H-2, J = 7.6 Hz); 5.53 (d, 1H, CH2Ph, J = 14.7 Hz); 6.47 (broad s, 1H, NH); 7.06–7.45 (m, 10H, Ph). Anal. Calcd. for C19H21N3O2: C, 70.57; H, 6.55; N, 12.99; Found: C, 70.87; H, 6.42; N, 12.52.
(±)-Trans-1-benzyl-6-oxo-2-phenylpiperidin-3-carbonylazide (12). To a stirred and cooled (0 °C) mixture of hydrazide 11 (0,323 g, 1 mmol), water (2.8 mL) and 38% hydrochloric acid (0.75 mL), a solution of NaNO2 (0.200 g, 3 mmol) in water (0.3 mL) was slowly added. After that the reaction mixture was stirred 15 min at 0 °C and extracted with dichloromethane. The organic solution was dried (Na2SO4) and the solvent was evaporated at reduced pressure to give 0.240 g (72%) azide 12 as an oily product. IR (Nujol): 1620 and 1705 (С = О), 2130 (N3) cm−1.
Methyl (±)-trans-1-benzyl-6-oxo-2-phenylpiperidin-3-ylcarbamate (9) via Curtius rearrangement. A solution of azide 12 (0.167 g, 0.5 mmol) in dry methanol (3 mL) was refluxed for 5 h and then the solvent was evaporated at reduced pressure. The residue was dissolved in dichloromethane (40 mL) and washed successively with 2.5% HCl, 10% Na2CO3 and water. The organic solution was dried (Na2SO4) and the solvent was evaporated. Column chromatography with light petroleum:ethyl acetate (2:1, v/v) of the residue yielded 0.236 g (72%) of 9 as white amorphous product, identical in all respects with methyl carbamate 9, prepared via Hofmann protocol.
General procedure for the synthesis of acylated derivatives of (±)-trans-5-amino-1-benzyl-6-phenylpiperidin-2-one (13–16). Solution of amine 7 (0.280 g, 1 mmol) and corresponding N-protected amino acid (1 mmol) in dichloromethane (5 mL) was cooled to -15 °C. DCC (1.3 mmol) was added portionwise and the reaction mixture was stirred for 1 h until completion of the reaction (tlc). The precipitated 1,3-dicyclohexylurea was filtered and filtrate was evaporated. Resulting oily residue was dissolved in ethyl acetate (50 mL) and washed with 1:1 HCl, water, 10% Na2CO3 and water. Organic phase was dried (Na2SO4) and the solvent was removed. Crude products were purified by means of column chromatography and/or recrystallization.
Benzyl 2-((±)-trans-1-benzyl-6-oxo-2-phenylpiperidin-3-ylamino)-2-oxoethylcarbamate (13). The compound was synthesized from amine 7 and Cbz-Glycine. Repeated recrystallization from ethyl acetate/light petroleum yielded 0.199 g (42%), mp 161–163 °C; IR (Nujol): 1620, 1670 and 1720 (С = О), 3250 and 3315 (NH) cm−1; 1H NMR (DMSO-d6): δ 1.43–1.93 (m, 2H, H-4), 2.51–2.62 (m, 1H, H-5), 2.68–2.89 (m, 1H, H-5), 3.22–3.57 (m, 2H, CH2Ph, CH2NH), 3.69 (dd, 1H, CH2NH, J = 5.9, 16.8 Hz), 3.90–4.02 (m, 1H, H-3), 4.48 (d, 1H, H-2, J = 3.1 Hz), 5.03 (s, 2H, OCH2Ph), 5.32 (d, 1H, CH2Ph, J = 15.4 Hz), 6.87–7.57 (m, 16H, NHCOO, Ph), 8.26 (d, 1H, NHCO, J = 7.0 Hz). Anal. Calcd. for C28H29N3O4: C, 71.32; H, 6.20; N, 8.91; Found: C, 71.70; H, 6.38; N, 9.20.
(2S)-N-((±)-trans-1-benzyl-6-oxo-2-phenylpiperidin-3-yl)-3-phenyl-2-(2,2,2-trifluoroacetamido)propanamide (14). The compound was synthesized from amine 7 and N-trifluroacetyl L-α-phenylalanine. Repeated recrystallization from ethyl acetate yielded 0.161 g (31%), mp 221–223 °C; IR (Nujol): 1625, 1665 and 1715 (C = O), 3225 (NH) cm−1; 1H NMR (DMSO-d6): δ 1.43–1.85 (m, 4H, 2xH-4, 2xH-4*), 2.51–3.03 (m, 8H, 2xH-5, 2xH-5*, 2xCHCH2Ph, 2xCHCH2Ph*), 3.26 (d, 1H, CH2Ph, J = 14.7 Hz), 3.36 (d, 1H, CH2Ph*, J = 14.7 Hz), 3.87–3.99 (m, 1H, H-3), 3.99–4.10 (m, 1H, H-3*), 4.44 (d, 1H, H-2, J = 3.8 Hz), 4.48 (d, 1H, H-2*, J = 3.6 Hz), 4.59 (dt, 1H, CHNH, J = 5.2, 9.1 Hz), 4.78 (dt, 1H, CHNH*, J = 5.0, 9.6 Hz), 5.30 (d, 1H, CH2Ph, J = 14.7 Hz), 5.36 (d, 1H, CH2Ph*, J = 14.7 Hz), 6.97–7.52 (m, 30H, Ph, Ph*), 8.56 (d, 1H, NHCO, J = 7.3 Hz), 8.63 (d, 1H, NHCO*, J = 7.1 Hz), 9.56 (d, 1H, NHCOCF3, J = 9.1 Hz), 9.60 (d, 1H, NHCOCF3*, J = 8.8 Hz). Anal. Calcd. for C29H28F3N3O3: C, 66.53; H, 5.39; N, 8.03; Found: C, 67.14; H, 5.31; N, 8.20.
Tert-butyl (2S)-1-((±)-trans-1-benzyl-6-oxo-2-phenylpiperidin-3-ylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-ylcarbamate (15). The compound was synthesized from amine 7 and Boc-L-tryptophan. Repeated recrystallization from ethyl acetate yielded 0.285 g (50%) mp 206–208 °C; IR (Nujol): 1610, 1655 and 1685 (С = О), 3310 and 3425 (NH) cm−1; 1H NMR (DMSO-d6): δ 1.28 (s, 9H, C(CH3)3), 1.33 (s, 9H, C(CH3)3*), 1.40–1.93 (m, 4H, 2xH-4, 2xH-4*), 2.53–2.98 (m, 8H, 2xH-5, 2xH-5*, 2xCH2Ind, 2xCH2Ind*), 3.28–3.45 (m, 2H, CH2Ph, CH2Ph*), 3.83–3.95 (m, 1H, H-3), 3.95–4.09 (m, 1H, H-3*), 4.14–4.37 (m, 2H, CHNH, CHNH*), 4.46 (d, 2H, H-2, H-2*, J = 3.2 Hz), 5.29 (d, 2H, CH2Ph, CH2Ph*, J = 15.1 Hz), 6.53 (d, 1H, indolic H, J = 8.6 Hz), 6.70 (d, 1H, indolic H*, J = 8.6 Hz), 6.90–7.67 (m, 30H, Ph, Ph*, NHCO, NHCO*), 8.14 (d, 1H, NHCOO, J = 7.5 Hz), 8.27 (d, 1H, NHCOO*, J = 7.1 Hz), 10.79 (s, 2H, indolic NH, indolic NH*). Anal. Calcd. for C34H38N4O4: C, 72.06; H, 6.76; N, 9.89; Found: C, 72.54; H, 6.72; N, 9.99.
Tert-butyl (2S)-1-((±)-trans-1-benzyl-6-oxo-2-phenylpiperidin-3-ylamino)-1-oxopropan-2-ylcarbamate (16). The compound was synthesized from amine 7 and Boc-L-α-alanine. Column chromatography with light petroleum:ethyl acetate (1:1, v/v) yielded 0.275 g (61%) of 16 as amorphous product, mp 95–98 °C; IR (Nujol): 1625, 1665 and 1705 (С = О), 3300 and 3420 (NH) cm−1; 1H NMR (CDCl3): δ 1.10 (d, 3H, CHCH3, J = 7.0 Hz), 1.18 (d, 3H, CHCH3*, J = 7.1 Hz), 1.44 (s, 9H, C(CH3)3), 1.50 (s, 9H, C(CH3)3*), 1.56–1.76 (m, 2H, H-4, H-4*), 1.82–2.01 (m, 2H, H-4, H-4*), 2.49–2.71 (m, 4H, 2xH-5, 2xH-5*), 3.30 (d, 1H, CH2Ph, J = 14.5 Hz), 3.32 (d, 1H, CH2Ph*, J = 14.5 Hz), 3.86–4.21 (m, 4H, H-3, H-3*, CHCH3, CHCH3*), 4.52 (s, 1H, H-2), 4.54 (d, 1H, NHCO, J = 7.7 Hz), 4.63 (s, 1H, H-2*), 4.93 (d, 1H, NHCO*, J = 7.7 Hz), 5.67 (d, 2H, CH2Ph, CH2Ph*, J = 14.5 Hz), 6.88 (br. s, 2H, NHCOO, NHCOO*), 7.16–7.52 (m, 20H, Ph, Ph*). Anal. Calcd. for C26H33N3O4: C, 69.16; H, 7.37; Found: C, 69.04; H, 7.30.
Conflict of interest statement
The authors state that there is no conflict of interest.
Acknowledgment
The financial support of the University of Sofia Science Fund (project 142/2007), National Science Fund of Bulgaria at the Ministry of Education and Science (project TK-X-1706/07) and project UNION (DO-02-82/2008) is gratefully acknowledged.