

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Polyoxometalate (POM) compounds, often described as soluble discrete metal oxides, can be finely designed at the molecular level. Because of their stunning compositions, diversified architectures, and fascinating topologies, POMs and their derivatives constitute a highly versatile class of compounds rich of more than several thousand species. This great diversity associated to a relatively easy synthetic pathway render them very attractive from fundamental point of view and for numerous properties or applications in many domains such as supramolecular chemistry [1, 2, 3], materials science [4, 5, 6], medicine [7, 8], magnetism [9, 10, 11, 12, 13], optics [11, 14], catalysis [15, 16, 17], or electrocatalysis [11, 18, 19].
Among these domains, the property that probably stands out is their unique electrochemical redox behavior, which can be finely tuned on purpose by changing their composition or their structures. In particular, with a tremendously diverse variety of POMs capable of storing a high number of electrons and displaying proton transfer capabilities, this class of compounds behaves as very efficient electrocatalysts for reactions of environmental interest such as CO2 reduction [20, 21, 22], proton reduction into hydrogen [23, 24, 25], oxygen evolution reaction [26, 27, 28, 29], or detection of environmental pollutants like nitrogen oxides [30, 31, 32, 33, 34, 35], bromates [35, 36, 37, 38], or iodates [35, 37, 39, 40, 41].
In our group, based on the fact that the incorporation of Mo- or W-based sulfurated clusters within POMs architectures is expected to modify their dynamic and electronic properties, and therefore their electrochemical and electrocatalytic properties, a fruitful approach for the syntheses for sulfur-containing polyoxometalates has been developed by combining the electrophilic {Mo2O2S2}2+ or {Mo3S4}4+ clusters with vacant polyoxotungstate ions or molybdates [42, 43, 44]. Some spectacular compounds have been obtained by following this strategy. However, the domain of sulfur-containing POMs has been neglected for a long time and the literature describing the electrochemical properties of such POMs is consequently very poor, while that of {Mo2O2S2}2+- or {Mo3S4}4+-based compounds exhibit very interesting properties for electrocatalytic reduction of protons into hydrogen in organic or in aqueous media [45, 46, 47, 48, 49, 50, 51, 52, 53].
In a recent paper, we studied the electrochemical properties of three compounds resulting from the combination of the divacant POM 𝛾-[SiW10O36]8− (denoted hereafter SiW10) with three different cationic building blocks, i.e.,
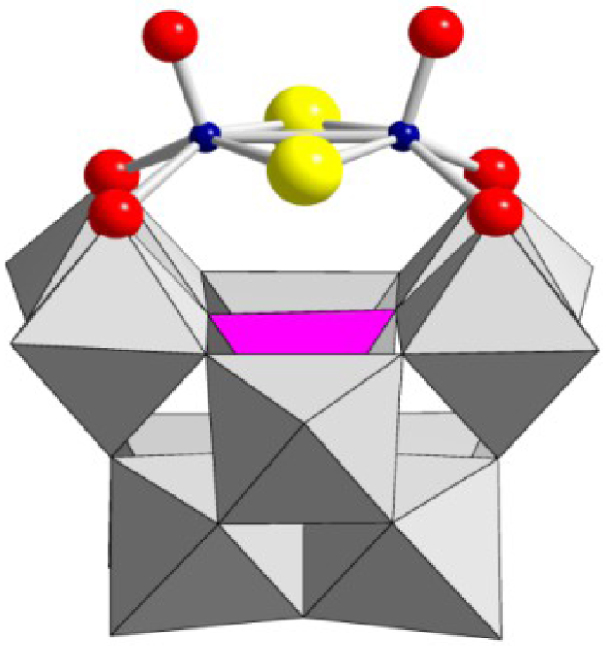
Structure of compounds [SiW10O36 (M2O2E2)]6− used in this study with M = Mo or W (blue spheres) and E = O or S (yellow spheres). Oxygen atoms are depicted in red, WO6 and SiO4 polyedra are given in gray and pink, respectively.
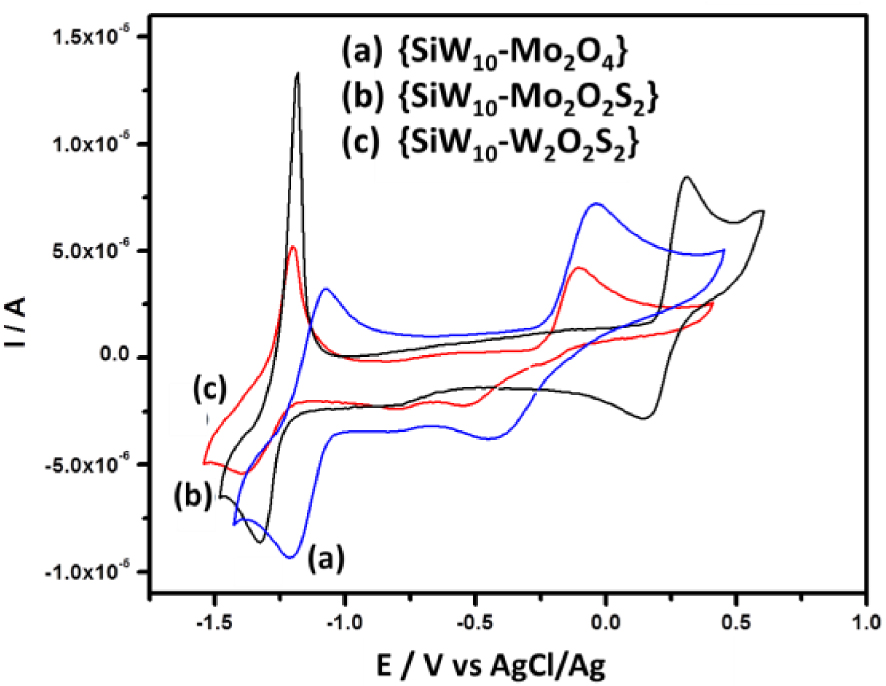
Cyclic voltammograms at a glassy carbon electrode (v = 0.1 V ⋅s−1) for 0.3 mM solutions of the three compounds {SiW10−Mo2O4} (a), {SiW10−Mo2O2S2} (b), and {SiW10−W2O2S2} (c) in dry DMF 0.1 M LiClO4 as the supporting electrolyte.
In this contribution, we decided to fill this work by exploring the electrochemical properties of these three compounds in aqueous medium and by investigating their potentialities in terms of electrocatalysis. A first part of this work is thus focused on the electrochemical properties of these molecular systems in sulfate buffers in comparison with results obtained in DMF. A second part is then focused on their electrocatalytic properties for the reduction of protons and different pollutants, and especially the reduction of iodate anions. In a last part, the most efficient compound of the series was immobilized on the surface of a glassy carbon electrode thanks to a layer-by-layer method [28, 32, 55, 56] using alternatively cetyltrimethylammonium bromide, CTAB, the POMs, and chitosan, a cationic biopolymer. The resulting modified electrode was then used for electroanalysis of iodate anions.
2. Materials and methods
2.1. Chemicals
Chemicals purchased from Aldrich Chemicals or Acros Chemicals were used without further purification. All solvents were of reagent grade quality and used without further purification. DMF was purchased from Sigma-Aldrich (anhydrous, purity 99.8%) and LiClO4 from Aldrich (purity 99.99%). The three compounds Cs6[SiW10O36(Mo2O2S2)]⋅6H2O (denoted hereafter {SiW10−Mo2O2S2}), Cs6[SiW10O36 (Mo2O4)]⋅6H2O (noted {SiW10−Mo2O4}), and Cs4.7K1.3 [SiW10O36(W2O2S2)]⋅7H2O (noted {SiW10−W2O2S2}) were prepared as previously described [57, 58] and characterized by usual routine methods (FT-IR, EDX, TGA).
2.2. Physical methods
Electrochemistry. Cyclic voltammetric (CV) experiments were carried out in buffer aqueous solution with an Autolab PGSTAT12 potentiostat/galvanostat associated with a GPES electrochemical analysis system (EcoChemie). Measurements were taken at room temperature in a conventional single compartment cell with an Ag/AgCl reference electrode, platinum gauze of large surface area, and a static surface modified glassy carbon disk working electrode. The source, mounting, and polishing of the glassy carbon electrodes (ø 3 mm) has been described previously [59]. The solutions were deaerated thoroughly for at least 30 minutes with pure argon and kept under a positive pressure of this gas during the experiments. Electrocatalytic reduction of iodates was studied by stepwise additions of potassium iodate solution in water. The surface modified electrode was prepared as follows: 5 μL of CTAB (30 g⋅L−1 in water) was dropped on a bare glassy carbon electrode and allowed to dry at room temperature, 5 μL of POM (2.5 g⋅L−1 in water) was then dried on the surfactant film. To avoid compounds released during experiment, a film of chitosan was then deposited. To do this, 3 μL of chitosan (1.5 g⋅L−1 prepared in acetic buffer) was dropped on the glassy carbon electrode and allowed to dry at room temperature.
3. Results and discussion
3.1. Electrochemical properties of {SiW10−Mo2O4}, {SiW10−Mo2O2S2}, and {SiW10−W2O2S2} in aqueous sulfate buffers
The cyclic voltammetry of 0.2 mM solution of {SiW10−Mo2O4}, {SiW10−Mo2O2S2}, and {SiW10−W2O2S2} in 0.5 M of sulfate buffer in the 1.5–3.7 pH range is presented in Figure 3, while Figure 4 and Figures S1–S4 in Supporting Information compare the CVs recorded for the three compounds at pH = 3 and pH = 1.5, respectively. Potentials are first scanned in the negative direction down to −1.0 V versus Ag/AgCl before going back in the direction of positive values up to + 0.8 V versus Ag/AgCl. Note that, as exemplified in Figures S2 and S3 for {SiW10−Mo2O4} and {SiW10−Mo2O2S2} (Supporting Information), the variation of the current of the first cathodic process are found proportional to the square root of the scan rate from 10 to 200 mV ⋅s−1, which indicates the electron exchanges are diffusion-controlled in these experimental conditions.
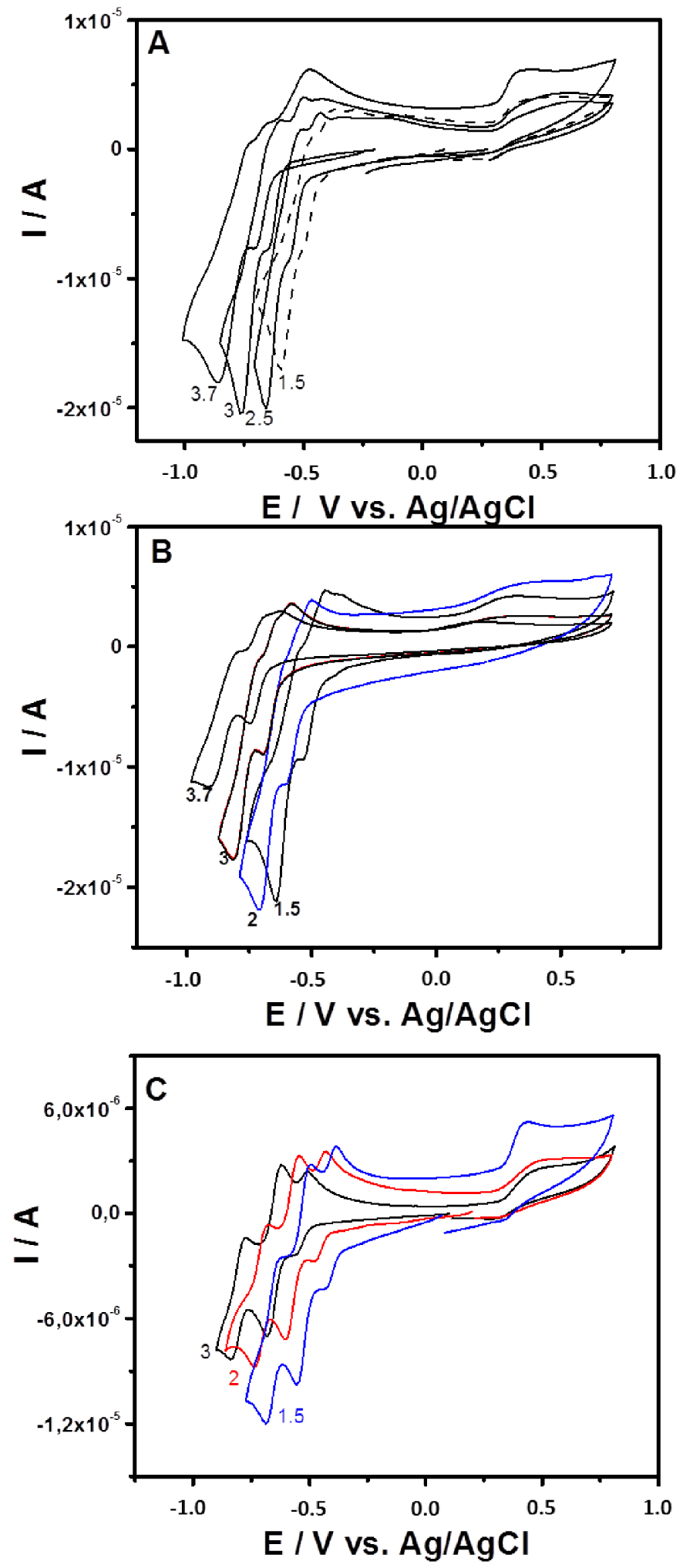
Cyclic voltammograms recorded at a glassy carbon electrode (v = 0.1 V ⋅s−1) in 0.1 M H2SO4∕Li2SO4 solutions containing (A) {SiW10−Mo2O4}, (B) {SiW10−Mo2O2S2}, and (C) {SiW10−W2O2S2} at 0.2 mM. The pH values are indicated on the corresponding CV.
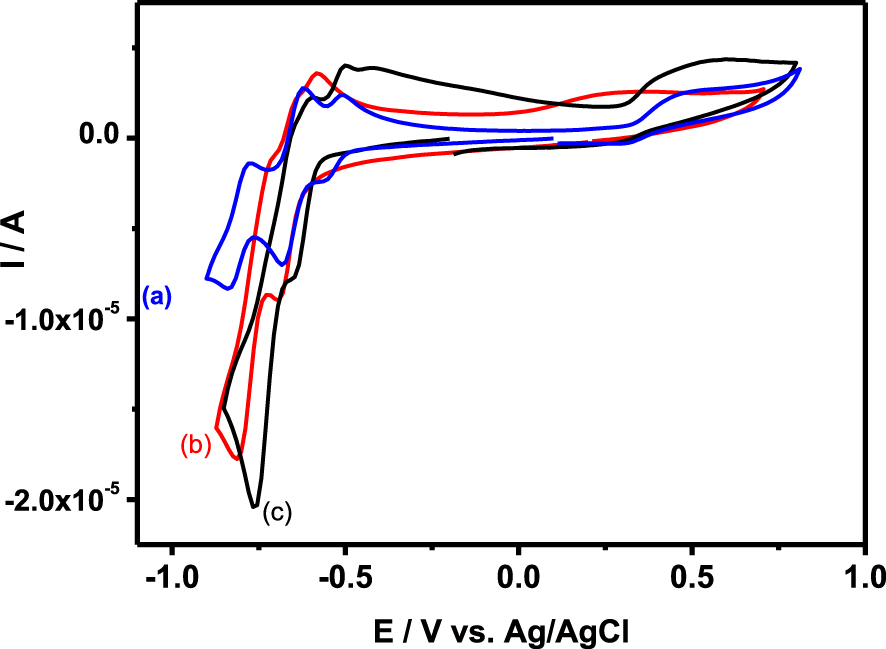
Cyclic voltammograms recorded at a glassy carbon electrode (v = 0.1 V ⋅s−1) in 0.1 M H2SO4∕Li2SO4 at pH = 3 for sulfate buffer solutions containing 0.2 mM {SiW10−W2O2S2} (a), {SiW10−Mo2O2S2} (b), and {SiW10−Mo2O4} (c).
Surprisingly, relatively few examples of electrochemical studies of SiW10-based POMs exist in the literature. The electrochemical behavior of the precursor [SiW10O36]8− and the derivative compounds [SiW10O36(PhPO)2]4− or [SiW10V 2O40]6− in aqueous acetate or sulfate buffers display well-defined reversible bi-electronic reduction waves within the −0.3 to −0.8 V versus Ag/AgCl range assigned to the reduction of four WVI centers of the SiW10 part. Furthermore, the latter are coupled with proton transfers, which render them pH-dependent [32, 60, 61]. In the case of the two “sandwich” compounds [(SiW10O36)2(Cr(OH)(H2O))3]10− and [(SiW10O36)2(Ru4O4(OH)2(H2O)4)]10−, the situation is relatively similar. For the former, in sulfate buffer, we can decompose the reduction of the SiW10 part into two main processes corresponding each to four electrons, two for each SiW10 moieties [62]. For the latter, the studies have been mainly focused on the oxidation part due to the capability of the Ru cluster to oxidize water into oxygen, but the reduction of the two SiW10 moieties displays at least one four electron process around −0.4 V versus Ag/AgCl in acidic aqueous medium [63, 64].
The CV obtained with {SiW10−Mo2O2S2} at pH = 3 exhibits as do the [SiW10O36]8− precursor, two reduction potentials at −0.670 V and −0.780 V (Figure 3B). On reversal potential, two reoxidation processes are observed, at −0.793 V and −0.637 V versus Ag/AgCl. The presence of an oxidation shoulder at ca −0.682 V indicates the composite nature of the second reduction wave. These waves correspond to the redox processes of the WVI in the polyoxoanion, which are consistent with those of reported sandwich-type POM [(SiW10O36)2(Cr(OH)(H2O))3]10− [62]. When the reversal potential is run up to 0.8 V, another oxidation peak appears at + 0.265 V. According to previous studies performed in DMF, this oxidation process is assigned to the oxidation of the two MoV centers of the [Mo2O2S2]2+ core into MoVI. This result suggests that the breaking of the Mo–Mo bond resulting from this oxidation process leads to a degradation of this compound in aqueous medium whatever the pH used in the present study. In contrast, this process was demonstrated to be relatively fast and chemically reversible in DMF medium (see Figure 2) [54].
In general, the reduction of POMs in aqueous medium is accompanied by proton transfers, which is translated by a pH-dependent electrochemical behavior. As expected, for the three compounds of this study, the reduction waves associated to the reduction of the SiW10 part move in the positive direction as the pH decreases over the pH range 1.5–3.7 in sulfate buffer (Figure 3), while the oxidation process of the clusters [M2O2E2]2+ (M = MoV or WV, E = S or O) appears not affected.
Compared to {SiW10−Mo2O2S2}, the cyclic voltammogram of {SiW10−Mo2O4} is very similar within the potential range explored (Figure 3A). At pH = 3, there is a positive potential shift of 80 mV and 130 mV for the first and second reduction steps, respectively (see Figure 4). As observed in DMF (see Figure 2), it shows that the reduction of SiW10 block in {SiW10−Mo2O4} is easier than the one in the sulfurated counterpart. A positive shift of irreversible oxidation wave was also observed and was found to be about + 0.479 V for the [Mo2O4]2+ cluster. This contrasts with the results obtained in DMF which shows a much more difficult oxidation for the [Mo2O2S2]2+ cluster.
Finally, Figure 3(C) shows the cyclic voltammogram of {SiW10−W2O2S2} in the same conditions. At pH = 3, at a more negative potential as do {SiW10−Mo2O4} and {SiW10−Mo2O2S2}, the CV of {SiW10−W2O2S2} exhibits three well-resolved reduction waves corresponding to the reduction of WVI atoms located at −0.536 V, −0.651 V, and −0.806 V, respectively, and the corresponding potential peak separation were calculated to be 57 mV, 65 mV, and 69 mV. As for {SiW10−Mo2O4} and {SiW10−Mo2O2S2}, at a more positive potential an irreversible oxidation peak corresponding to the oxidation of WV in the [W2O2S2]2+ core was observed at + 0.511 V. As can be seen from Figure S4 (SI), the plots of E°′ of the three redox waves (denoted I, II, and III) versus pH show all good linearity in the pH range. The slopes of the three redox couples in this pH range are−84 mV/pH (I),−84 mV/pH (II), and−85 mV/pH (III). The slope values corresponding to these three redox processes are slightly higher compared to the theoretical value of−59 mV/pH for the 2e−∕2H+ redox process. However, there were evidences of higher protonation requirement. For {SiW10−Mo2O4} and {SiW10−Mo2O2S2}, the plots of Epc corresponding to the reduction of W centers of SiW10 in these compounds versus pH also show good linearity in the same pH range with 40 and 46 mV/pH as slope, respectively.
3.2. Electrocatalytic properties in aqueous sulfate buffer medium
As mentioned in the introduction, POMs can be exploited extensively in electrocatalytic reduction due to their ability of delivering electrons and protons to other species. It is noteworthy to mention that the three studied POMs did not show any significant catalytic activity toward nitrates, nitrites, and bromates, whereas reduction of protons probably slightly occurs. In contrast, the reduction of iodate appears efficient. The efficiency of {SiW10−Mo2O4}, {SiW10−Mo2O2S2}, and {SiW10−W2O2S2} for the electrocatalytic reduction of iodate into iodide was first evaluated qualitatively in cyclic voltammetry experiments. The CVs depicted in Figure 5 were recorded at a bare glassy carbon electrode in the pH 3 medium and shows the following main observations in sequence: in each case the reduction waves of the three compounds are well-defined in the absence of
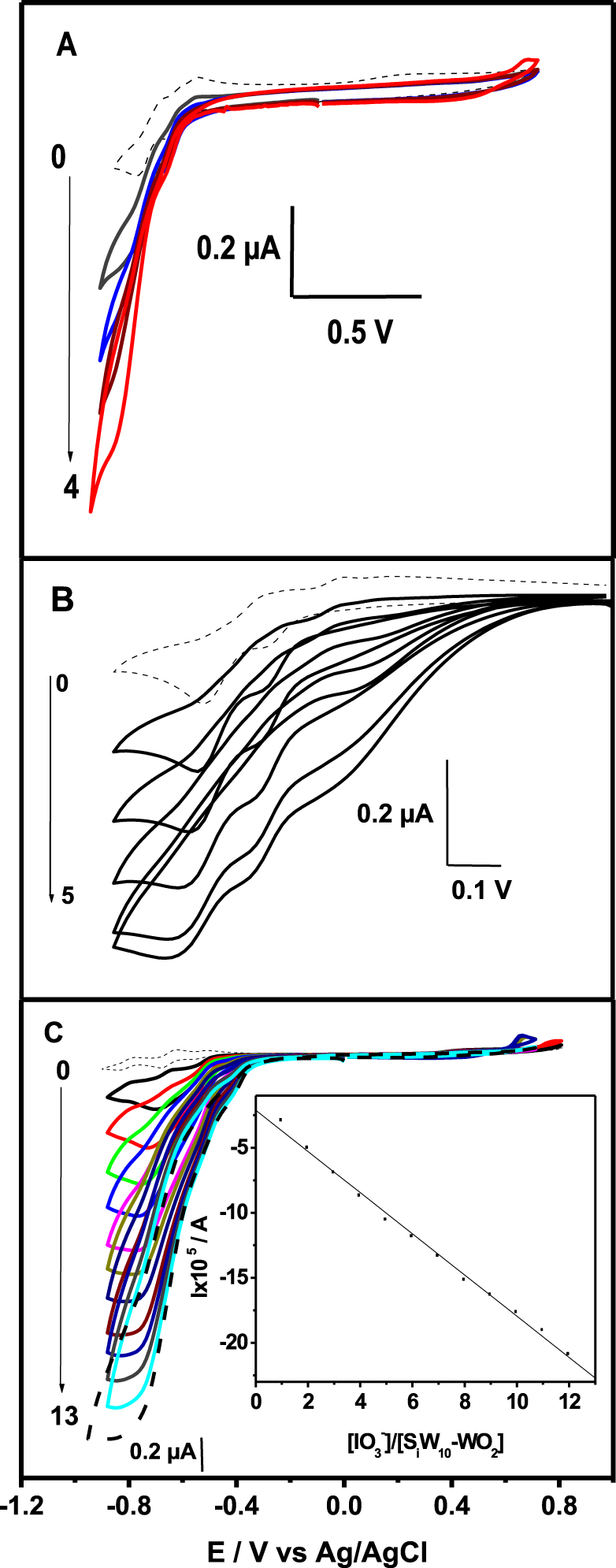
Cyclic voltammograms for the electrocatalytic reduction of iodate on a glassy carbon electrode by 0.2 mM {SiW10−Mo2O4} (A), {SiW10−Mo2O2S2} (B), and {SiW10−W2O2S2} (C) in pH 3 buffer (0.1 M Li2SO4 + H2SO4). Scan rate was 0.1 V ⋅s−1. Inset shows the influence of 𝛾 on the electrocatalytic current. Arrows on the left part of the figure indicate the range of the 𝛾 values.
When the catalytic current contains a component due to the reduction of the POM itself, the catalytic efficiency can be defined as:
3.3. Application to the electroanalysis of iodates
Immobilization of POMs remains a major point for their applications. Particular attention has been paid toward the immobilization of POMs on different surfaces to enhance their stability and achieve well-organized nanoassemblies with accessible redox states. The latter have proven, in many examples, their ability to be immobilized onto the surface of electrodes. Various techniques have been employed, but the layer-by-layer assembly method (LBL) probably appears to be the simplest and most effective method to develop well-ordered architectures with precisely controlled thicknesses resulting in films that exhibit excellent thermal, mechanical, and chemical stabilities [28, 32, 55, 56, 65].
{SiW10−W2O2S2} was immobilized on the glassy carbon electrode for iodate electroanalysis in aqueous medium by following an LBL methodology: on a clean polished glassy carbon electrode was deposited 5 μL of a solution of cetyltrimethylammonium bromide (CTAB, 30 g⋅L−1). After washing with water and drying in air at room temperature, 5 μL of POM (2.5 g⋅L−1) was dried on the surfactant film and then, to avoid the compound released during experiment, a film of chitosan, a cationic biopolymer, was then deposited. To do this, 3 μL of chitosan (1.5 g⋅L−1 prepared in acetic buffer) was dropped on the glassy carbon electrode and allowed to dry at room temperature for giving a modified electrode ideally corresponding to Scheme 1.
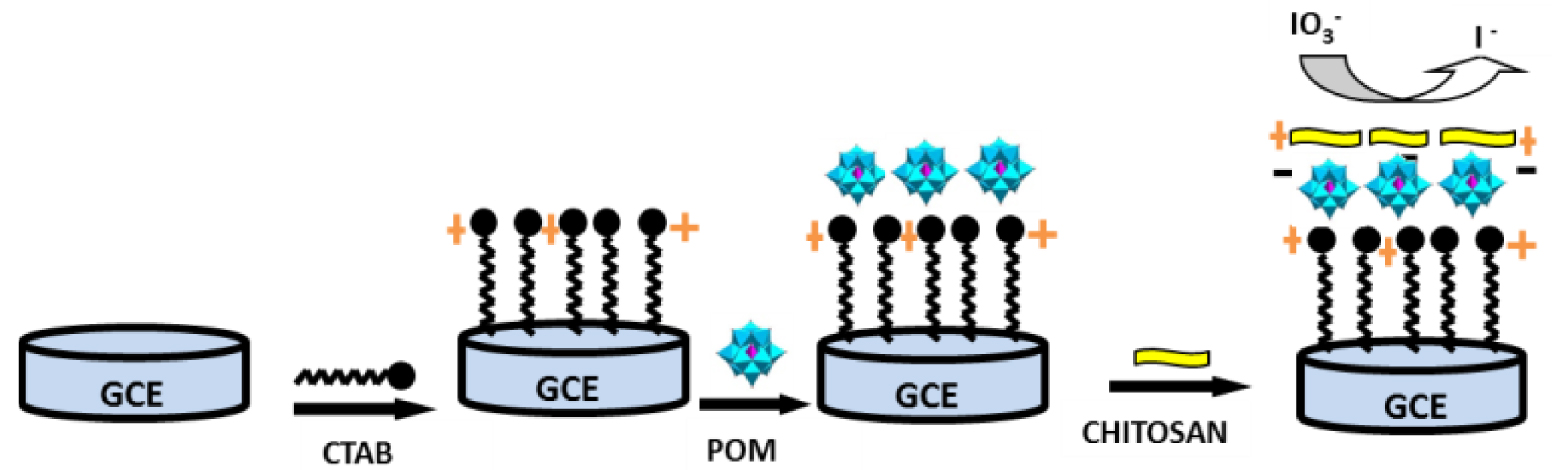
Idealized schematic view of the preparation of the modified electrode used in this study. Counterions such as bromides and alkali cations are omitted for clarity. Note that the organization of the deposit at the surface is probably more disorganized than this theoretical view.
Figure 6 shows the cyclic voltammograms recorded in argon saturated 0.1 M H2SO4∕Li2SO4 pH 2.5 aqueous buffer solution at GCE/CTA/{SiW10−− W2O2S2}/Chitosan, pH 2.5 being the optimal pH value determined in the 1.5–3.7 pH range to perform the studies. It can be seen from this that in the potential range −0.800 to + 0.800 V, three reversible or quasi-reversible redox waves appear and the difference of peak potential (𝛥E) are 214 mV for wave I, 58 mV for wave II, and 89 mV for wave III. The mean peaks potential E°′ are 219 mV (I), −396 mV (II), and −615 mV (III) versus AgCl/Ag, respectively. Since CTA+ cations and chitosan are not electroactive, their presence is not reflected in the cyclic voltammograms, which demonstrates that the electrochemical behavior of the {SiW10−W2O2S2} anions studied is maintained in the multilayer deposits. These redox processes are likely attributed to the consecutive reduction processes occurring at the tungsten atoms (WVI→WV) at a more negative potential and the oxidation of (WV→WVI) at a more positive potential accordingly to the previous studies. Interestingly, the oxidation process of the [W2O2S2]2+ cluster (wave I) appears quasi-reversible, as observed in DMF, which suggests that the oxidized compound is stabilized in such a matrix.
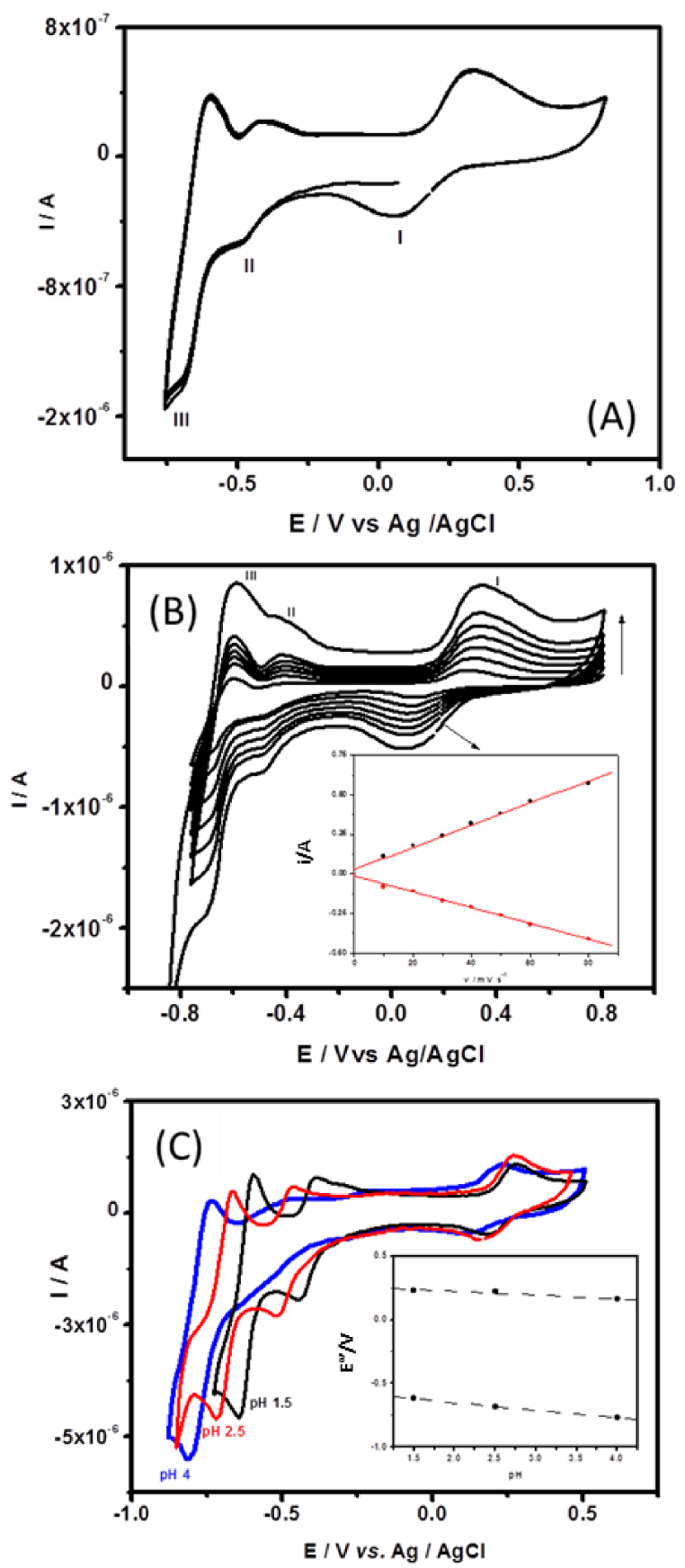
(A) Cyclic voltammograms of GCE/CTA/{SiW10−W2O2S2}/Chitosan in pH 2.5 buffer (0.1 M H2SO4 + 0.1 MLi2SO4) solution. Scan rate: 50 mV ⋅s−1. (B) Cyclic voltammograms of GCE/CTA/{SiW10−W2O2S2}/Chitosan in pH 2.5 buffer (0.1 M H2SO4 + 0.1 MLi2SO4) at different scan rates in the range of 10–80 mV ⋅s−1. Inset shows linear variation of currents versus scan rates. (C) Cyclic voltammograms recorded at GCE/CTA/ {SiW10−W2O2S2}/Chitosan 0.1 M H2SO4 + 0.1 M Li2SO4 solution at different pH (1.5–3.7), scan rate 75 mV ⋅s−1. Inset shows the relationship between peak potential and pH. Masquer
(A) Cyclic voltammograms of GCE/CTA/{SiW10−W2O2S2}/Chitosan in pH 2.5 buffer (0.1 M H2SO4 + 0.1 MLi2SO4) solution. Scan rate: 50 mV ⋅s−1. (B) Cyclic voltammograms of GCE/CTA/{SiW10−W2O2S2}/Chitosan in pH 2.5 buffer (0.1 M H2SO4 + 0.1 MLi2SO4) at different scan rates in the range of 10–80 mV ⋅s−1. Inset shows linear ... Lire la suite
Scan effect on the electrochemical behavior of GCE/CTA/{SiW10−W2O2S2}/Chitosan electrode was investigated in the potential range +800 to −800 mV. When the scan rates varied from 5 to 80 mV ⋅s−1, the cathodic currents are almost the same as the corresponding anodic current and the peak potential did not change much with increasing scan rate (Figure 6B). The plot of peak (I) current versus scan rates is shown in the inset of Figure 6(B). The peak currents show a linear dependence with scan rates up to 80 mV ⋅s−1, suggesting that the redox process is surface confined and that the charge transport in the film is fast [66, 67].
The pH of the solution has a remarkable effect on the electrochemical behavior of GCE/CTA/ {SiW10−W2O2S2}/Chitosan. As shown in Figure 6(C), it can be seen that along with increasing the pH (1.5–4), both anodic and cathodic peak potentials of waves I and II gradually shift to a more negative potential direction and the corresponding peak currents decreased. Although peak II at pH 4 is ill-defined, the plot of peak potentials of processes I and III versus pH show good linearity (see inset in Figure 6C). The slopes of these lines are about 59 mV/pH for wave III and only about 28 mV/pH for wave II accordingly to solution studies.
An important requirement of modified electrode for the sensor application is the stability in contacting solution or sample. Therefore, the stability of GCE-modified electrode was checked in 0.1 M sulfate buffer solutions at pH 2.5. By performing 10 successive cyclic voltammograms (Figure 6A), the modified electrode shows a relatively stable response in peak intensities and peak potentials of {SiW10−W2O2S2} immobilized at the surface of the electrode remain nearly unchanged, which indicates that the modified electrode can be used conveniently for sensor application.
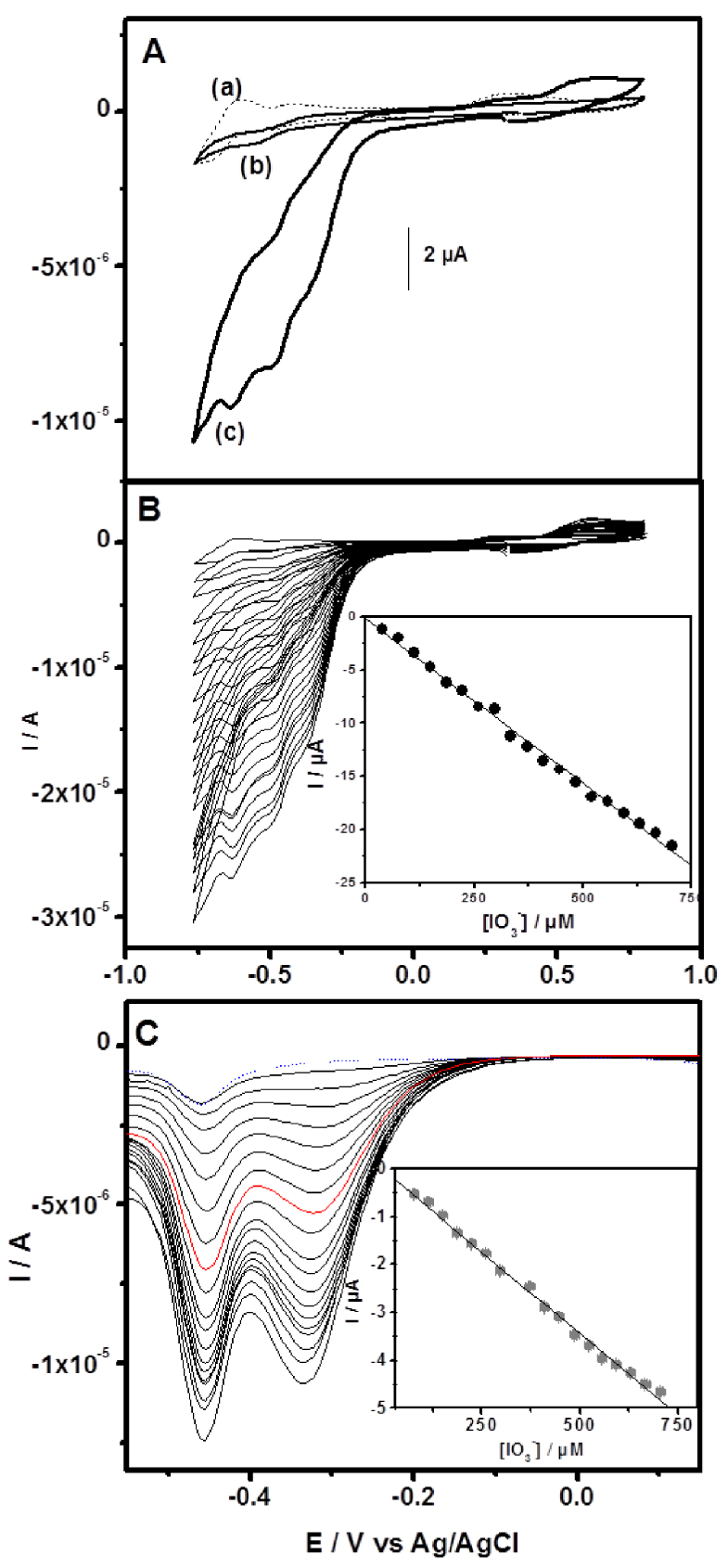
(A) Cyclic voltammograms recorded in sulfate buffer pH 2 in the absence of iodate at GCE/CTA/{SiW10−W2O2S2}/Chitosan (a) and in the presence of iodate at GCE/CTA/Chit (b) and GCE/CTA/{SiW10−W2O2S2}/Chitosan (c). (B) Cyclic voltammograms and (C) Square wave voltammograms recorded with GCE/CTA/ {SiW10−W2O2S2}/Chitosan for different concentration of iodate in pH 2.5 buffer. Inset is the iodate reduction current versus its concentration.
As it is known, the electroreduction of
This current was found to be linearly dependent on the iodate concentration from 37 μM to 709 μM with a correlation coefficient of 0.998 (inset Figure 7B). Square wave voltammetry responses of iodate on GCE/CTA/{SiW10−W2O2S2}/Chitosan electrode obtained by successively adding iodate to an electrochemical cell is shown in Figure 7(C), while the linear calibration plot of peak currents versus concentrations is given in inset. Based on the data shown in inset, it could be ascertained that the reduction currents of iodate at −0.336 V were linear with the concentration. The linear regression equation was calculated as Ip∕μA =−0.0345− 0.0068 c∕μM in the range of 74–705 μM (R2 = 0.998). Based on the signal-to-noise ratio of 3 (S∕N = 3), the detection limit was calculated as 6.2 μM. As prepared, GCE/CTA/ {SiW10−W2O2S2}/Chitosan electrode showed a reasonable sensitivity and a comparatively low detection limit. The analytical properties of proposed modified electrode are comparable to those of other previously reported chemically modified electrodes with POMs [68, 69, 70, 71].
4. Conclusions
In this paper, we studied the electrochemical behavior of three isoelectronic compounds [SiW10O36(M2O2E2)]6− (M = Mo or W; E = S or O) in aqueous acidic medium in comparison with previous studies performed in DMF [54]. The reduction processes centered on the SiW10 part are in agreement with other SiW10 derivatives of the literature. Interestingly, the oxidation of the (M2O2E2)2+ cluster associated to the breaking of the M–M bond appears almost independent from the pH and irreversible in aqueous medium while this process was quasi-reversible or reversible in DMF.
The electrocatalytic properties of these POMs were investigated in sulfate buffer. Only the electrocatalytic reduction of the iodates revealed to be efficient and [SiW10O36(W2O2S2)]6− is clearly the most efficient POM of the series. The latter was immobilized by a layer-by-layer technique onto the surface of a glassy carbon electrode. The electrochemical behavior of the POM is maintained in these conditions and the reversibility of the oxidation process lost in aqueous solution is recovered. The resulting modified electrode was tested for the electroanalysis of iodates in aqueous acidic medium, evidencing a limit detection value of 6.2 μM, which appears among the good results of the literature of POMs to our knowledge.