


 CC-BY 4.0
CC-BY 4.0
1. Introduction
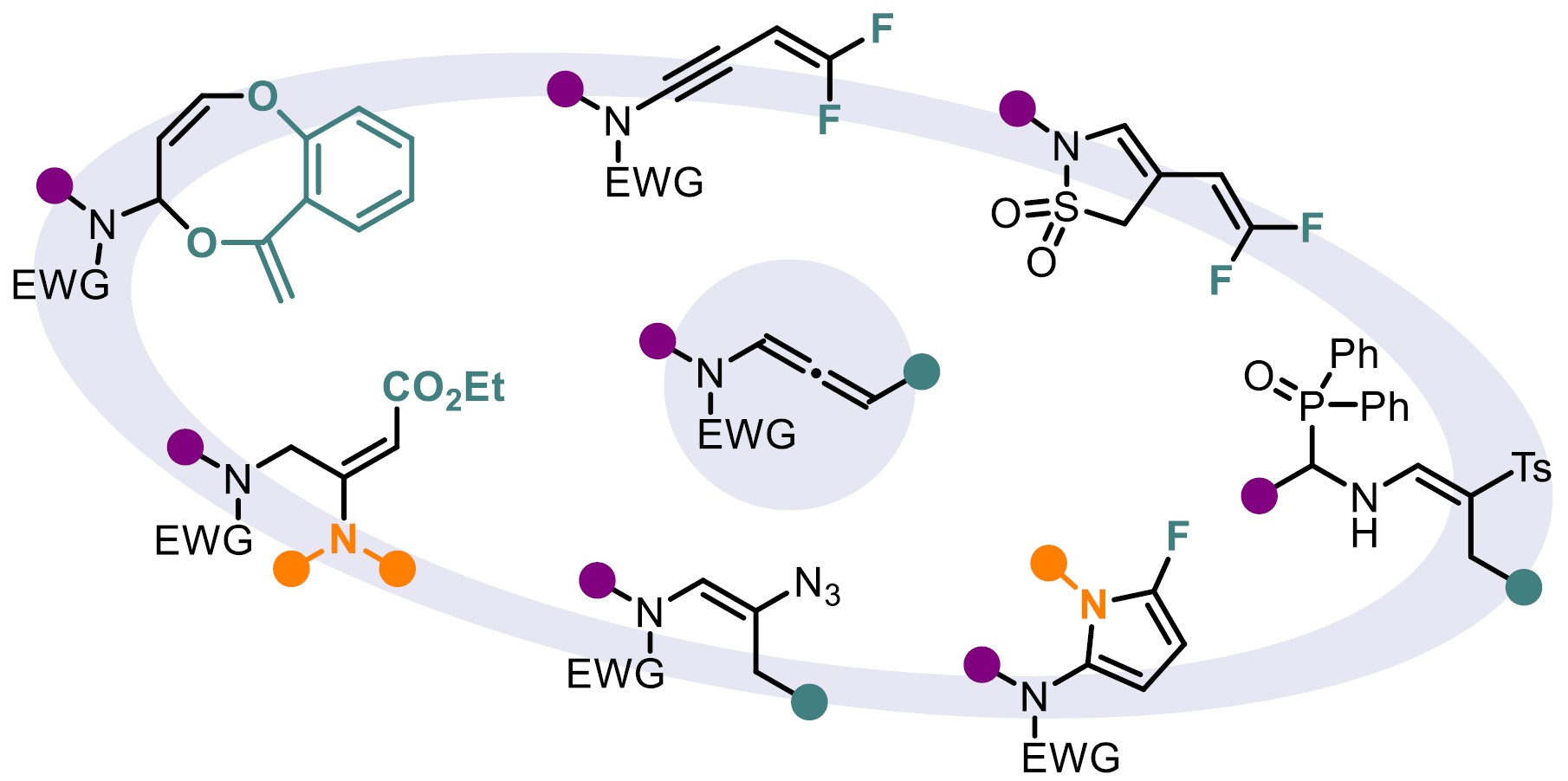
N-Allenamides, featuring an electron-withdrawing group (EWG) on the nitrogen atom, have gained significant attention among organic chemists over the past decade, surpassing their allenamine counterparts. This increased interest stems from their enhanced stability and retained distinctive reactivity. The delocalization of the nitrogen’s lone pair into the allenic system imparts these compounds with dual reactivity, as depicted in their resonance structures (Figure 1). As a consequence, a range of highly regio- and stereoselective functionalizations at the α-,β-, and γ-positions of the allene moiety has been developed. These transformations enable the construction of complex and valuable N-heterocycles, often yielding either proximal or distal adducts with precise control. The extensive focus that has been dedicated to N-allenamides is clearly reflected in the substantial number of publications and comprehensive reviews that have emerged over the years, highlighting their diverse and powerful reactivity as versatile synthetic building blocks (for selected reviews on N-allenamide, see [1, 2, 3, 4, 5]). Over the past three years, our research group has initiated a dedicated program focused on exploring the reactivity of activated N-allenamides. Our initial approach involved the use of ynamides as precursors to access these valuable intermediates. We then shifted our attention to investigating their reactivity, with particular emphasis on the construction of heterocyclic frameworks which are key structural motifs commonly found in natural products.
N-allenamide structure and reactivity profile.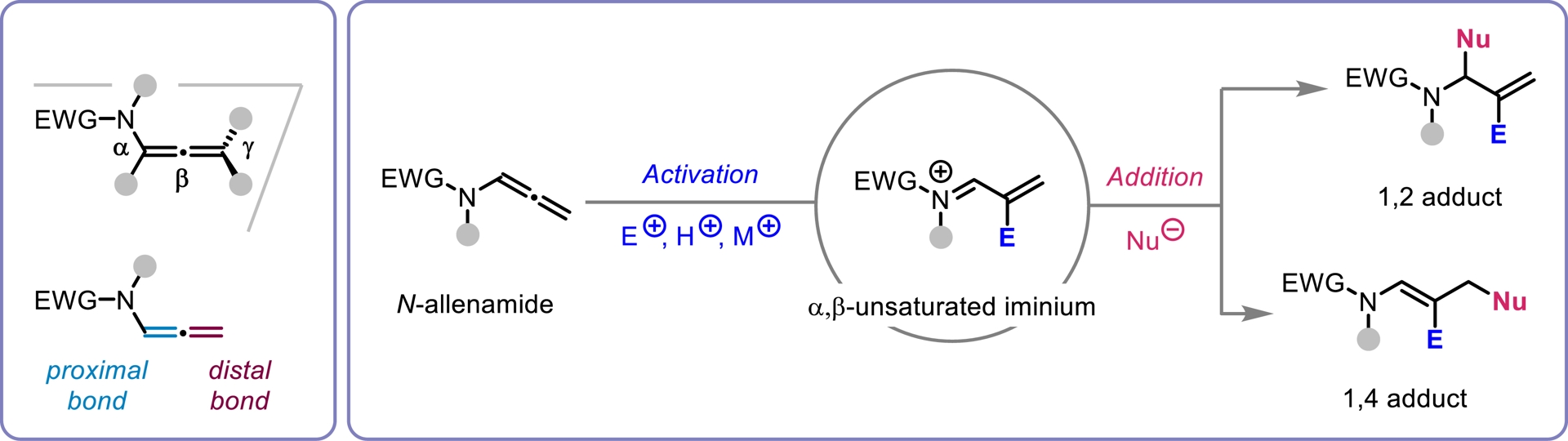
2. Results and discussions
N-Allenamides, a subclass of allenes, have emerged as essential motifs in a myriad of transformations. Despite their growing prominence as unique and versatile building blocks in organic synthesis, existing methods for their preparation often suffer from limited substrate scope and restricted structural diversity. Inspired by the addition of diazo compounds to alkynes [6, 7, 8, 9, 10], we envisioned that a coupling reaction between terminal ynamides and a readily available diazomethane surrogate, trimethylsilyldiazomethane (Me3SiCHN2, 2 M in diethyl ether), in the presence of tetra-n-butylammonium fluoride (TBAF) could offer a straightforward route to terminal N-allenamides. We were pleased to find that the desired coupling products were obtained in good yields, and the reaction exhibited broad functional group tolerance, such as fluorinated substituents (Scheme 1) [11].
Synthesis of terminal and fluorinated N-allenamides.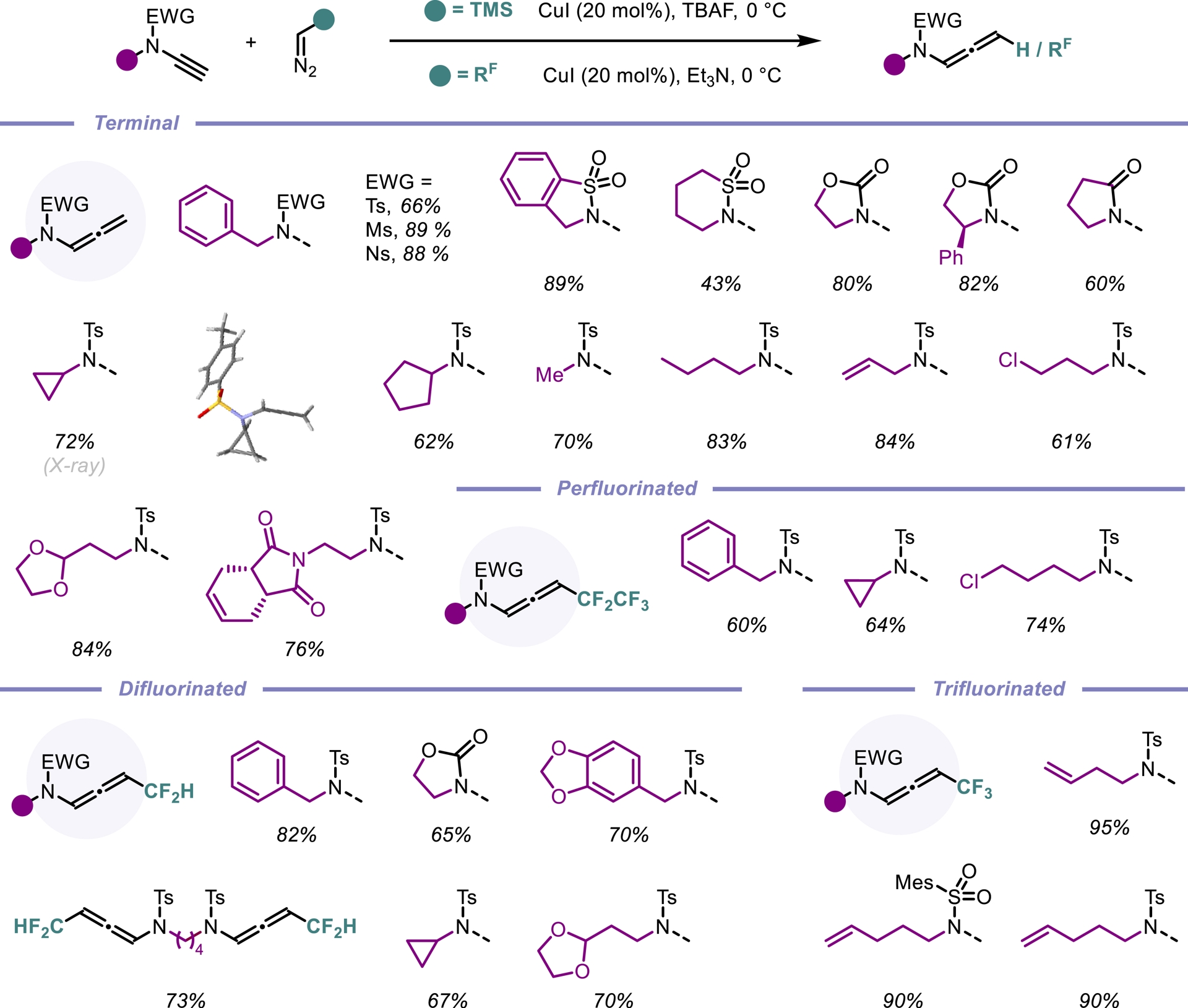
Fluorine-containing building blocks have become essential in the development of new drug candidates [12, 13, 14]. Despite major advances in fluorine chemistry, fluorine-substituted N-allenamides remain scarce [15]. Building on this, we sought to explore the synthesis of fluorinated N-allenamides using fluorinated diazoalkanes. Addition of trifluorodiazoethane to terminal ynamides under copper catalysis in acetonitrile led to the formation of the desired N-allenamides in very good yields. The pentafluoroethyl group, another highly fluorinated motif, also proved compatible with this transformation, cleanly delivering the corresponding N-allenamides. Encouraged by these results, we turned our attention to more challenging fluorinated substituents, notably the difluoromethyl group (CF2H). Recognized as a metabolically stable bioisostere of hydroxyl and thiol groups, the CF2H moiety has gained prominence in pharmaceutical, agrochemical, and materials science [16, 17]; for recent reviews on difluoromethylation reactions see [18, 19, 20]. When CF2HCHN2 was reacted with electron-deficient alkynes, we successfully obtained CF2H-substituted N-allenamides from terminal ynamides under similar conditions. Remarkably, the reaction displays broad functional group tolerance, accommodating oxazolidinones, halogens (e.g., chlorine), protected aldehydes, and even unsaturated side chains that offer further opportunities for downstream functionalization (Scheme 1).
Building on our efficient access to a new class of push–pull allenes, we turned our attention to hydroaminations, an atom-economical reaction for constructing diverse heterocyclic scaffolds, which serve as valuable platforms for further functionalization. While significant progress has been made in hydroaminations targeting the proximal C=C bond [21, 22] and, to a lesser extent, the distal C=C bond [23, 24], additions to the central C=C bond remain comparatively rare. To address this gap, we employed our ynamide/diazo coupling method to synthesize N-allenamides bearing an ester substituent. Although these intermediates were highly unstable, they proved amenable to hydroamination, yielding a variety of amido-captodative enamines. Notably, the activated N-allenamides underwent regio- and stereoselective addition with a range of secondary amines. A counterintuitive anti-Michael addition provides a unique E-configured enamine. Numerous amides as well as distinct amines could be adapted (Scheme 2). Density functional theory (DFT) calculations further supported the regio- and stereoselectivity of the reaction [25].
One-pot hydroamination of activated N-allenamides.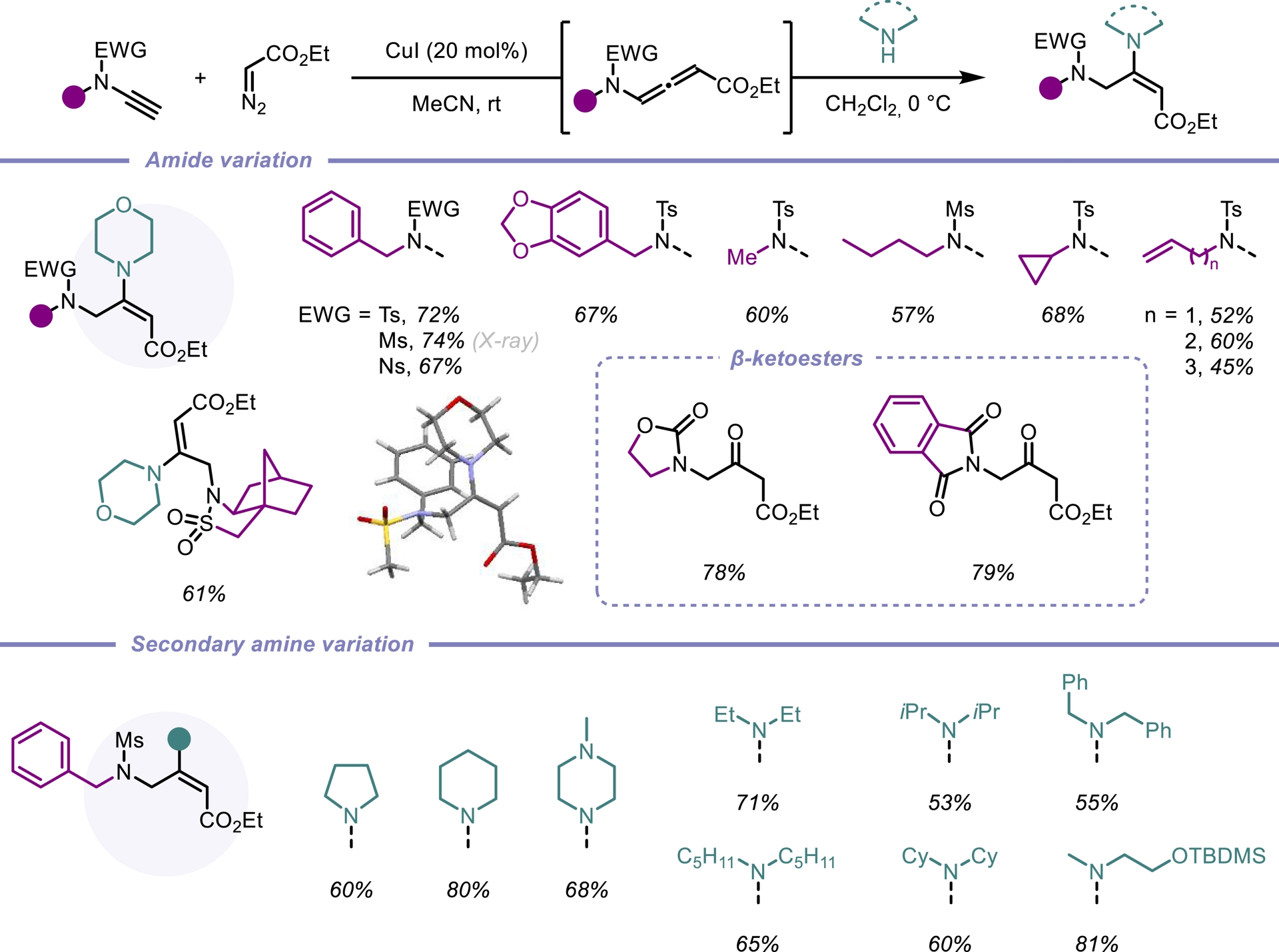
Eight-membered heterocycles are conspicuous structural motifs found in numerous natural products and/or bioactive molecules. However, their synthesis remains challenging due to unfavorable enthalpic and entropic barriers associated with the transition states required for medium-size ring formation [26, 27, 28]. Among these, benzo[b][1,5]-dioxocines stand out as particularly valuable eight-membered heterocycles. Notable examples include penicillide [29] and a derivative discovered by Bayer [30], both exhibiting significant biological activity. These compounds act as cholesteryl ester transfer protein (CETP) inhibitors and have emerged as promising candidates for the treatment of dyslipidemia (Figure 2) [31]. Reported Conia-ene reactions of ene-ynamides remain scarce [32, 33]. As part of our ongoing research on ynamide reactivity, we became interested in exploring the behavior of ynamides tethered to propargylic ethers under basic conditions [34]. Given that propargylic ethers are known to isomerize in the presence of a base, we hypothesized an enolate addition to the in-situ formed N-allenamide ether. Among the bases evaluated, a solution of benzyltrimethyl-ammonium hydroxide (Triton B) in methanol emerged as the optimal choice to perform this transformation. Experimental observations were supported by DFT calculations, indicating a regioselective O-alkylation, where the enolate adds to the proximal position of the N-allenamide. This transformation furnishes eight-membered rings featuring two embedded enols and a hemiaminal core. Importantly, the structure tolerates modifications at the terminal tether of the ynamide as well as both substituents on the nitrogen atom, demonstrating the versatility of this method in generating diverse, highly functionalized substructures. This approach offers a sustainable and green route to access shelf-stable eight-membered O-heterocycles, significantly expanding the accessible chemical space for this underexplored class of compounds (Scheme 3). The computed energy profiles show that the pathway involving the formation of an N-allenamide intermediate can be considered as the preferential one, essentially due to the lower activation energy and more favorable initial deprotonation step [35].
Bioactive benzo[b][1,5]dioxocines.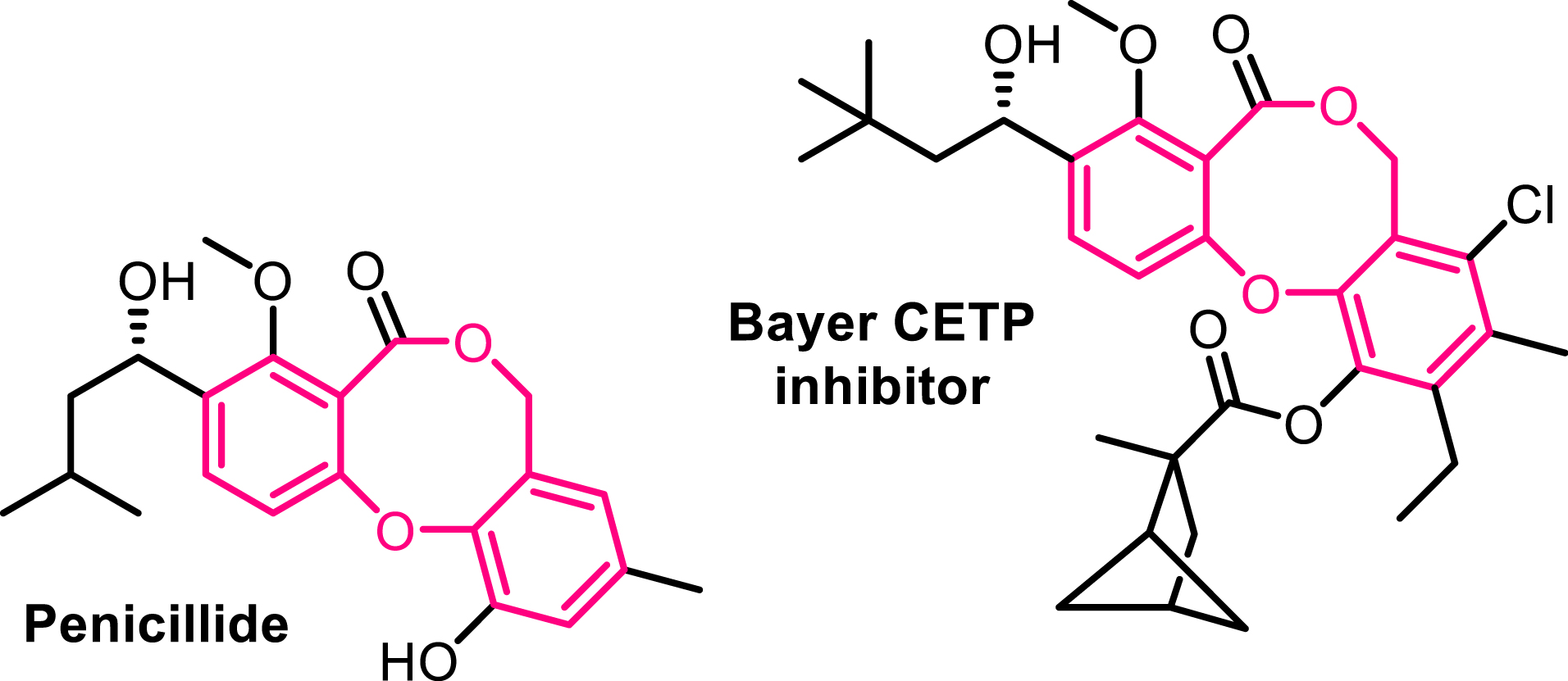
Construction of eight-membered O-heterocycles from tethered ynamides.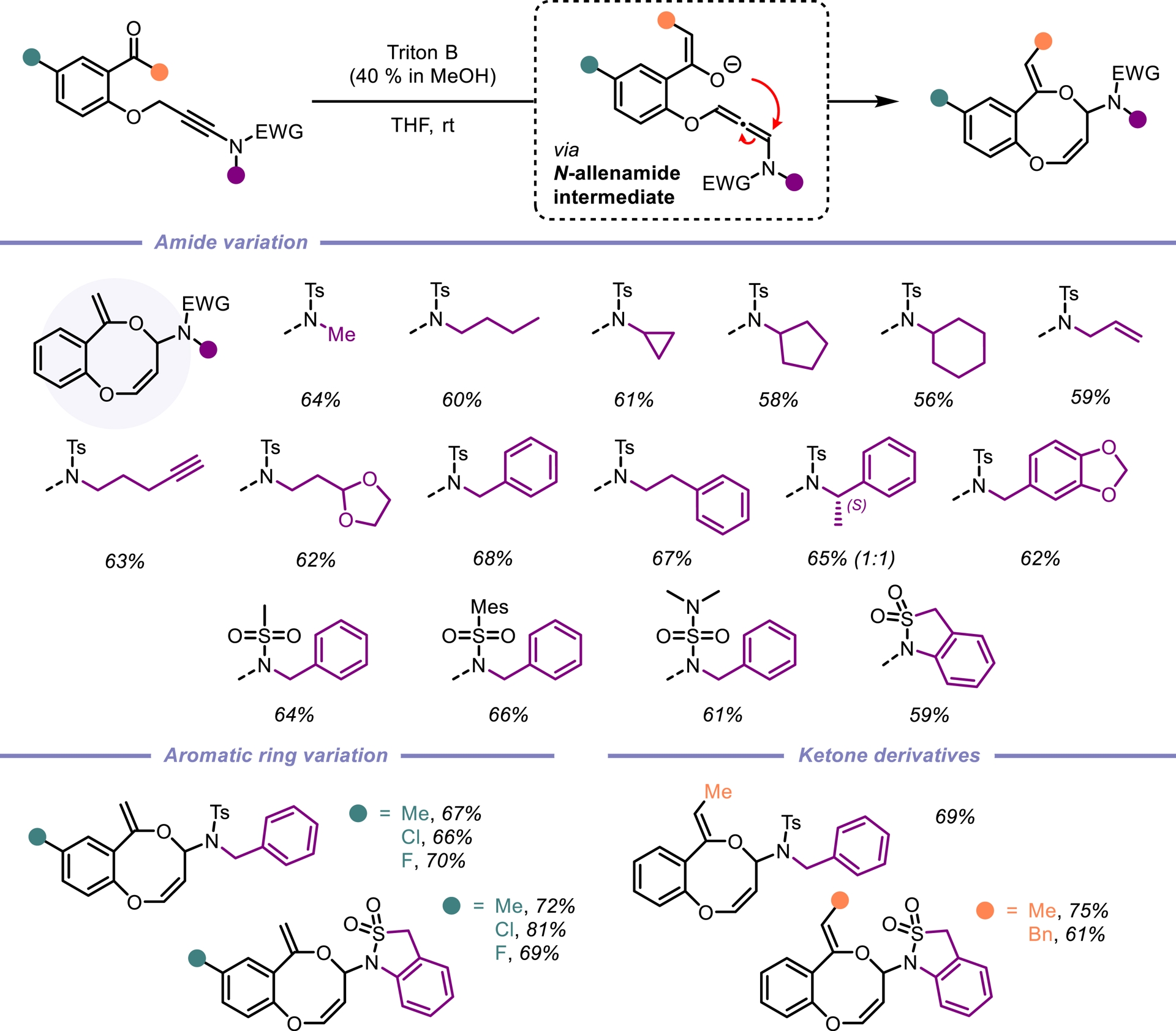
In light of our previous findings, we investigated whether trifluoromethylated N-allenamides could be leveraged to generate ene-ynamides with a particular emphasis on incorporating gem-difluoroalkene and monofluoroalkene motifs. These fluorinated alkenes are especially appealing: gem-difluoroalkenes act as carbonyl mimics, and monofluoroalkenes serve as bioisosteres of peptide and enol bonds [36]. For example, the pyrrolidone derivative seletracetam demonstrates a lack of psychomimetic effects, offering significant advantages over many conventional antiepileptic drugs [37]. In-vivo studies have shown that a difluoromethylated GABA aminotransferase inhibitor is highly effective in suppressing dopamine release following acute exposure to cocaine or nicotine [38]. The presence of a fluoroalkene transformed the opioid peptide Leu-enkephalin into an in-vivo probe with sufficient plasma and microsomal stability to provide an orally active peptide probe with central nervous system distribution (Figure 3) [39]. We found that a base-promoted δ-elimination of a fluorine atom from the CF3-substituted N-allenamides efficiently afforded gem-difluorinated ene-ynamides. Both potassium tert-butoxide (t-BuOK) and sodium hydride (NaH) proved to be effective bases for this transformation. The reaction proceeded smoothly and exhibited a broad functional group tolerance, accommodating a variety of functionalized side chains and electron-withdrawing groups, thus highlighting the versatility of this strategy (Scheme 4) [40]. Given the polarity of the ynamides and the high electrophilicity of the α-carbon of the gem-difluoroalkene moiety, it was possible to tune the reactivity of these peculiar building blocks by adding nucleophiles on the ynamide moiety or on the difluoroalkenyl part. Alcoholates were directly added to the starting N-allenamides. Interestingly, this reaction yielded a mixture of Z- and E-monofluorinated ene-ynamides, with the alkoxy group at the carbon atom δ to the nitrogen. The overall yield was good, and the two stereoisomers could be successfully separated, affording both Z- and E-configured ene-ynamides (Scheme 5). Subsequent selective syn-hydroacylation and hydro-chlorination promoted by ZrCl4 occurred exclusively at the ynamide moiety, enabling the synthesis of tailored mono- or difluorinated dienes depending on the starting material (Scheme 6) [40].
Bioactive mono- and difluorinated alkenes.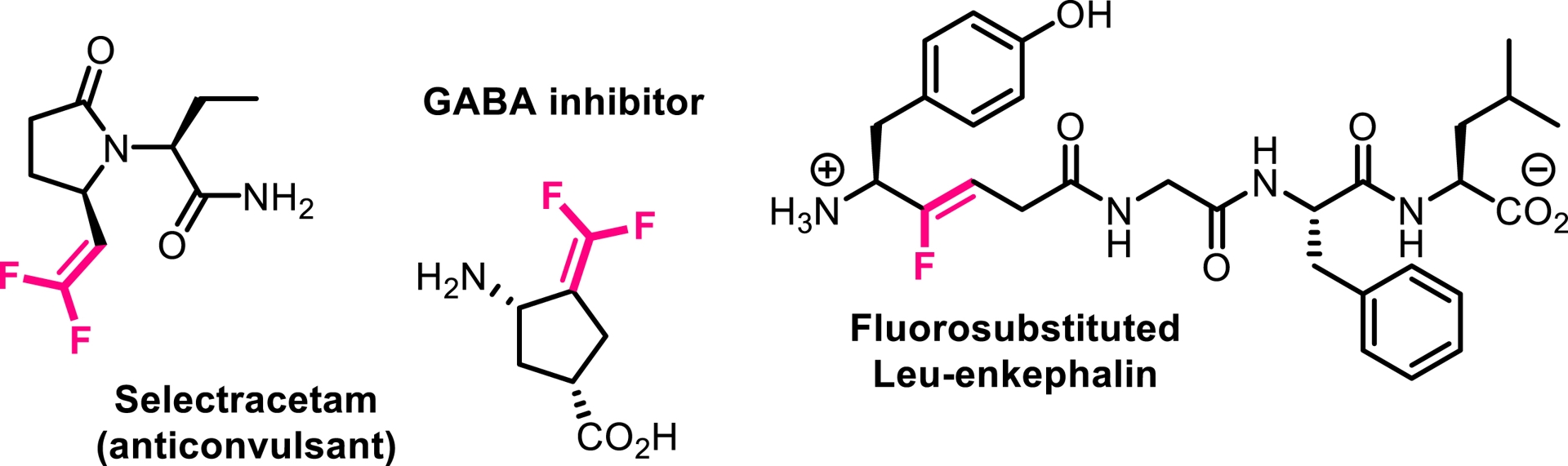
Synthesis of gem-difluorinated ene-ynamides.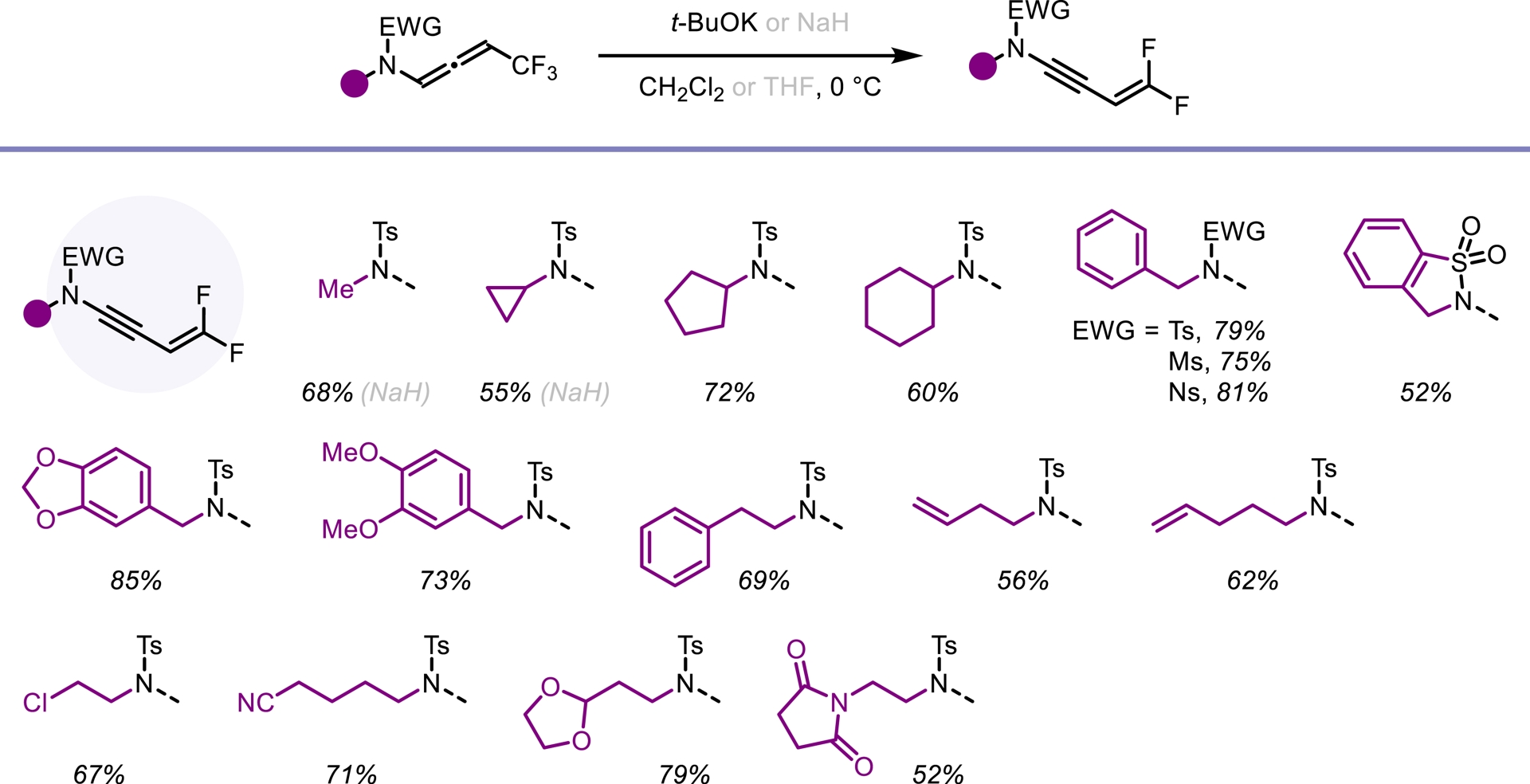
Regioselective substitution on the alkenyl moiety.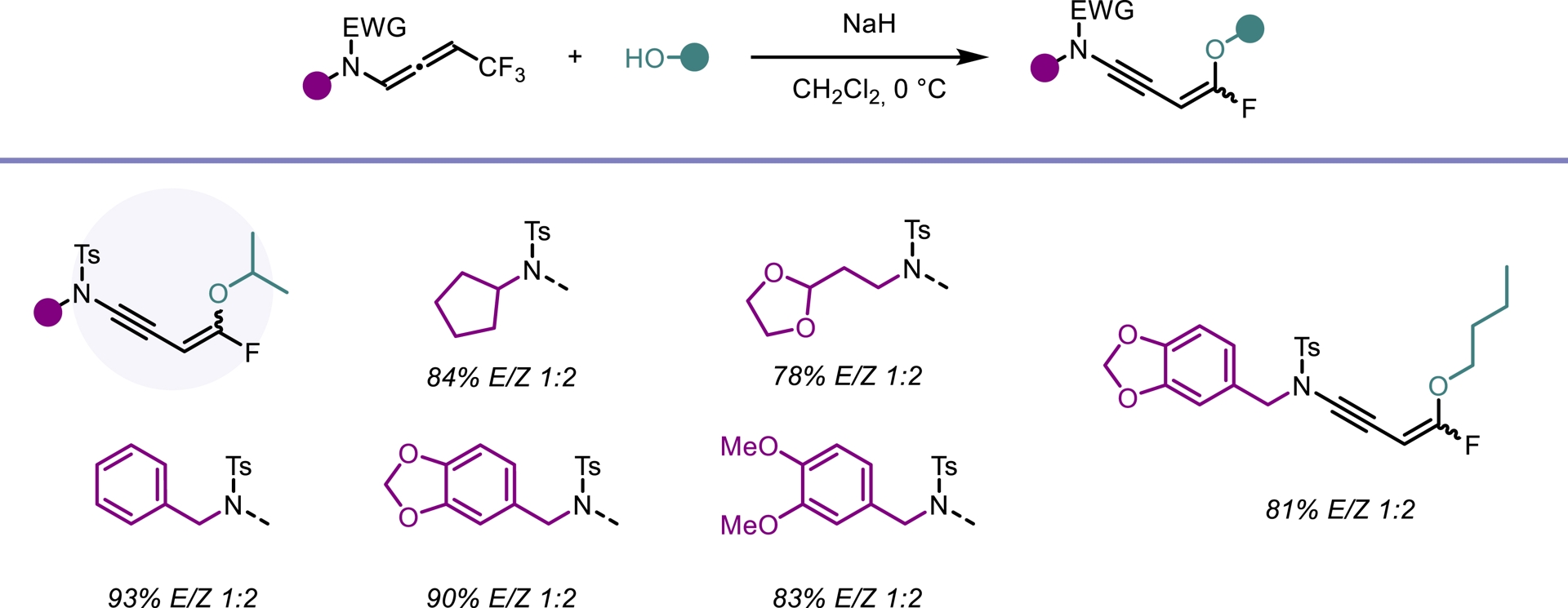
Hydrofunctionalization of the ynamide moiety.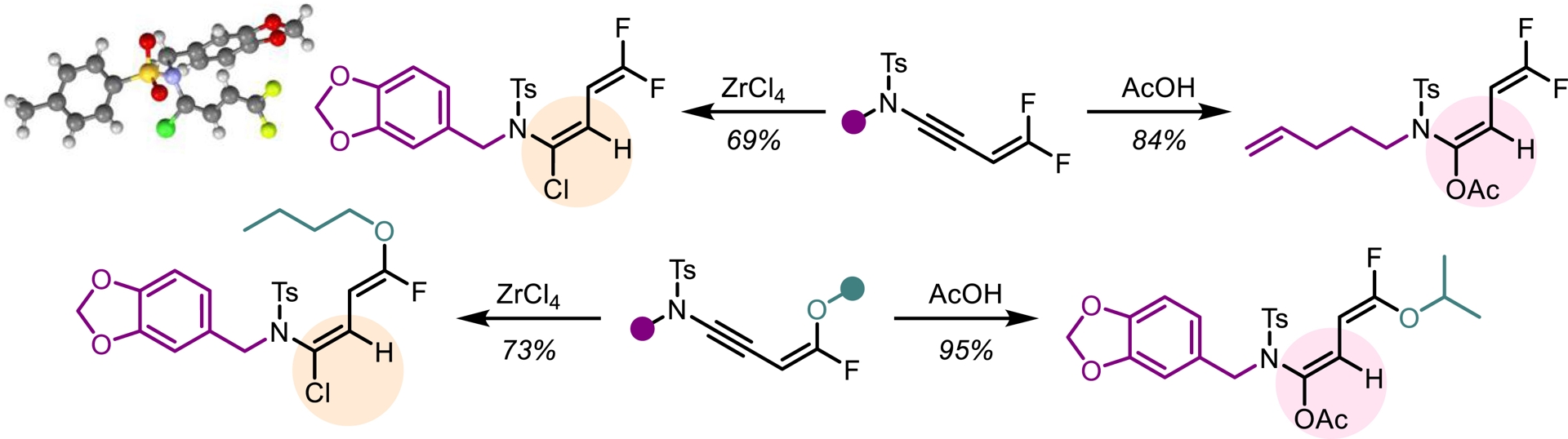
Despite their attractive properties, only a limited number of protocols have been developed for the synthesis of fluoropyrroles. Currently, fluorinated pyrroles are predominantly obtained through the functionalization of pre-existing pyrrole frameworks [41, 42, 43]. Building on our previous results, we envisioned that gem-difluorinated ene-ynamides [40] could serve as suitable precursors for the synthesis of fluorinated pyrroles via hydroamination followed by an intramolecular cyclization. To explore this possibility, we investigated the use of silver catalysts to promote the regioselective addition of primary amines to ene-ynamides. In contrast to alcoholates, primary amines selectively add to the ynamide moiety of the molecule generated in situ. Upon silver-mediated activation of the gem-difluoroalkenyl group, a 5-endo-trig cyclization followed by β-fluoride elimination occurred, furnishing the desired fluorinated pyrrole scaffold (Scheme 7). This transformation exhibits high modularity, allowing straightforward access to a wide range of highly functionalized 2-amido-5-fluoropyrroles. Good yields were obtained for this operationally simple process involving a one-pot four-step sequence (Scheme 8). An intramolecular version of this process led to the synthesis of tricyclic oxazoles from monofluorinated ene-ynamides embedding a primary amine (Scheme 9) [44].
Proposed reaction mechanism.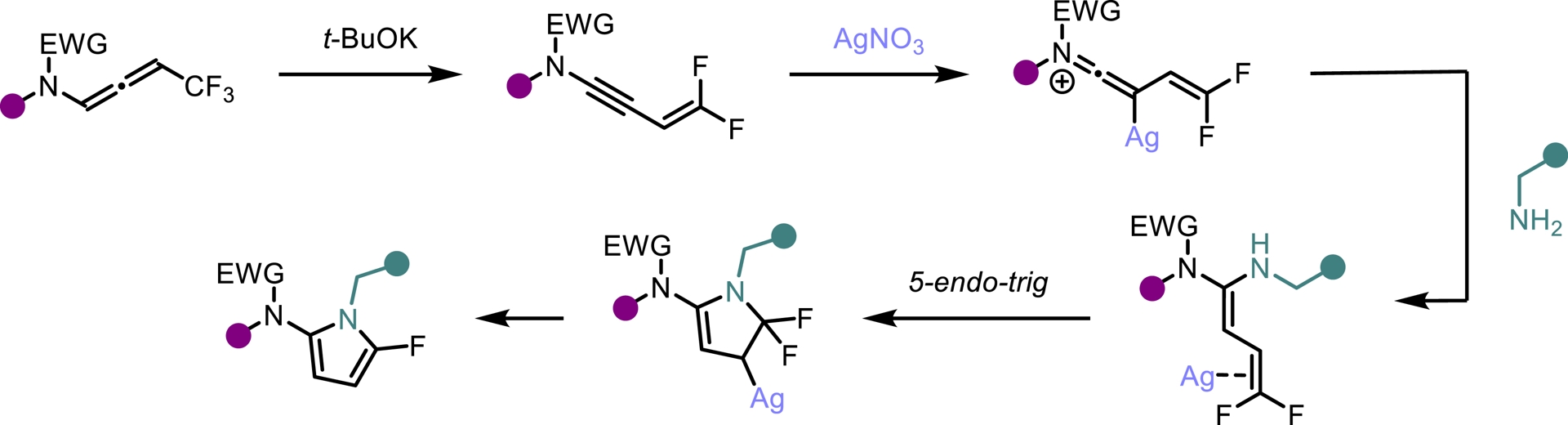
Domino synthesis of fluorinated pyrroles.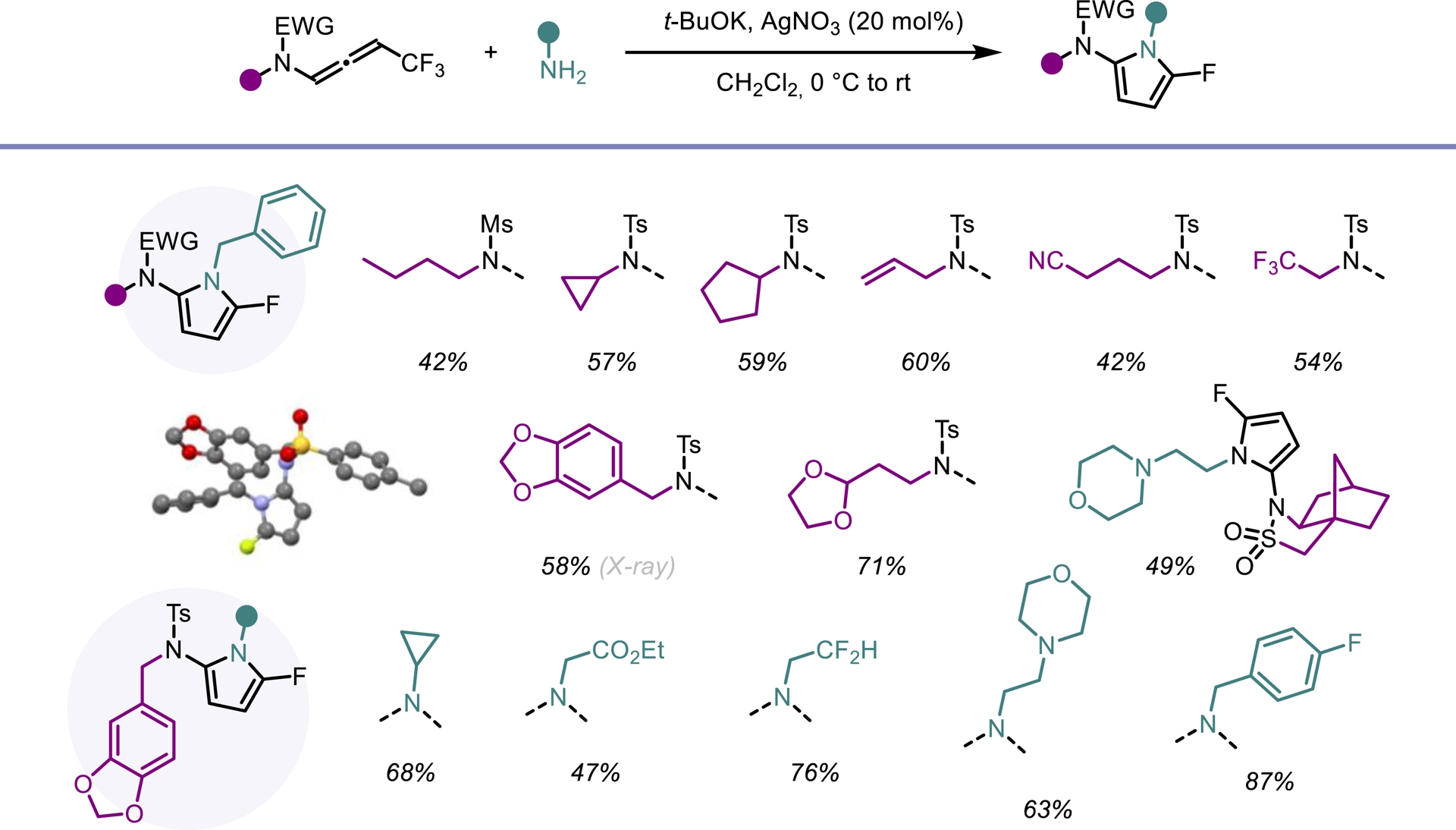
Synthesis of oxazoles from monofluorinated ene-ynamides.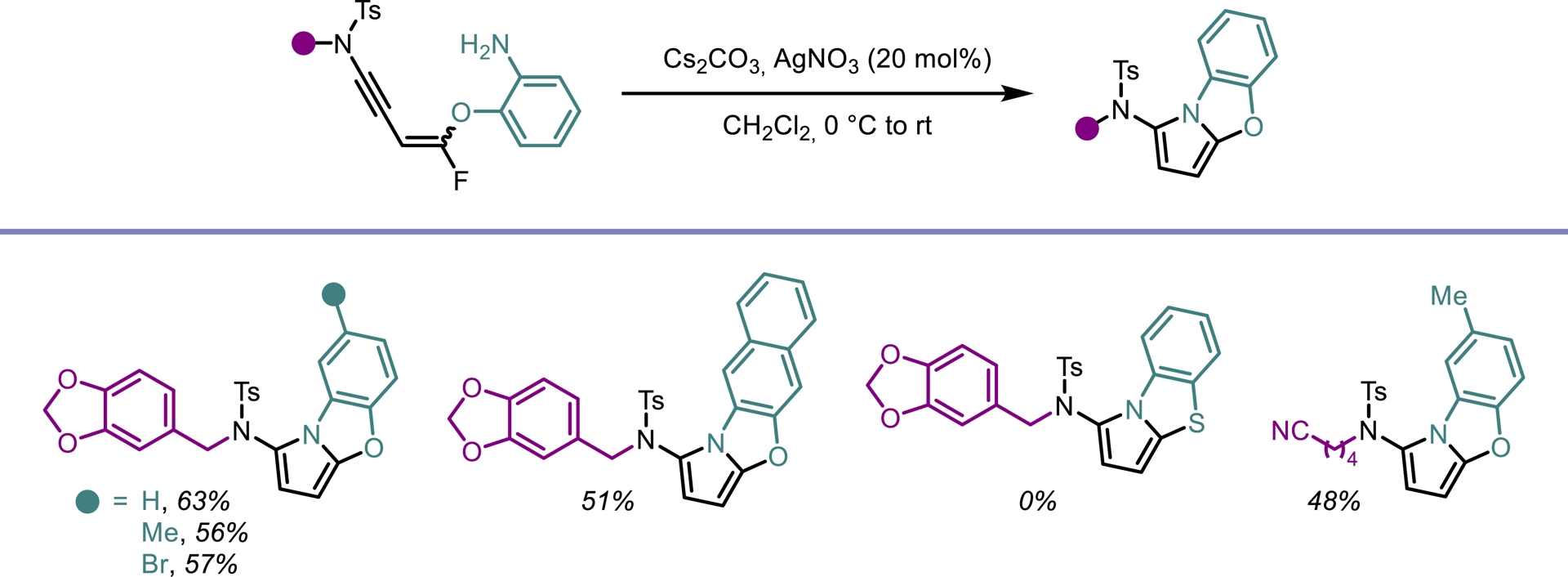
Since the discovery of sulfonamide antibacterials [45], sulfonamides have played a key role in medicinal chemistry. The cyclic counterparts of sulfonamide compounds (sultams) display enhanced biological activities compared to their acyclic congeners [46]. Our strategy aimed to exploit the electron-withdrawing substituent on the nitrogen atom in trifluorinated N-allenamides. Specifically, we hypothesized that deprotonation at the α-position of the sulfonyl group in N-sulfonyl allenamides could trigger an anionic 5-endo-dig cyclization, ultimately leading to the formation of cyclic sulfonamides. Although not found in nature, these amide surrogates are considered privileged motifs that have found diverse applications in drug discovery such as in sultiame, an anticonvulsant agent [47], S-2474, a non-steroidal anti-inflammatory drug [48], and an antidiabetes agent developed by Boehringer Ingelheim (Figure 4) [49]. Upon treatment with TBAF and acetic acid, CF3-substituted N-allenamides were transformed into γ-sultams bearing an ene-gem-difluorinated tether, whereas TBAF alone provided the corresponding trifluorinated ethyl sultams. The developed method demonstrated broad functional group tolerance and a wide substrate scope, affording sultams featuring a gem-difluorinated ene moiety (Scheme 10). Moreover, DFT calculations supported a reaction pathway involving a 5-endo-dig cyclization on the ene-ynamide intermediate generated in situ [50].
Bioactive sultams.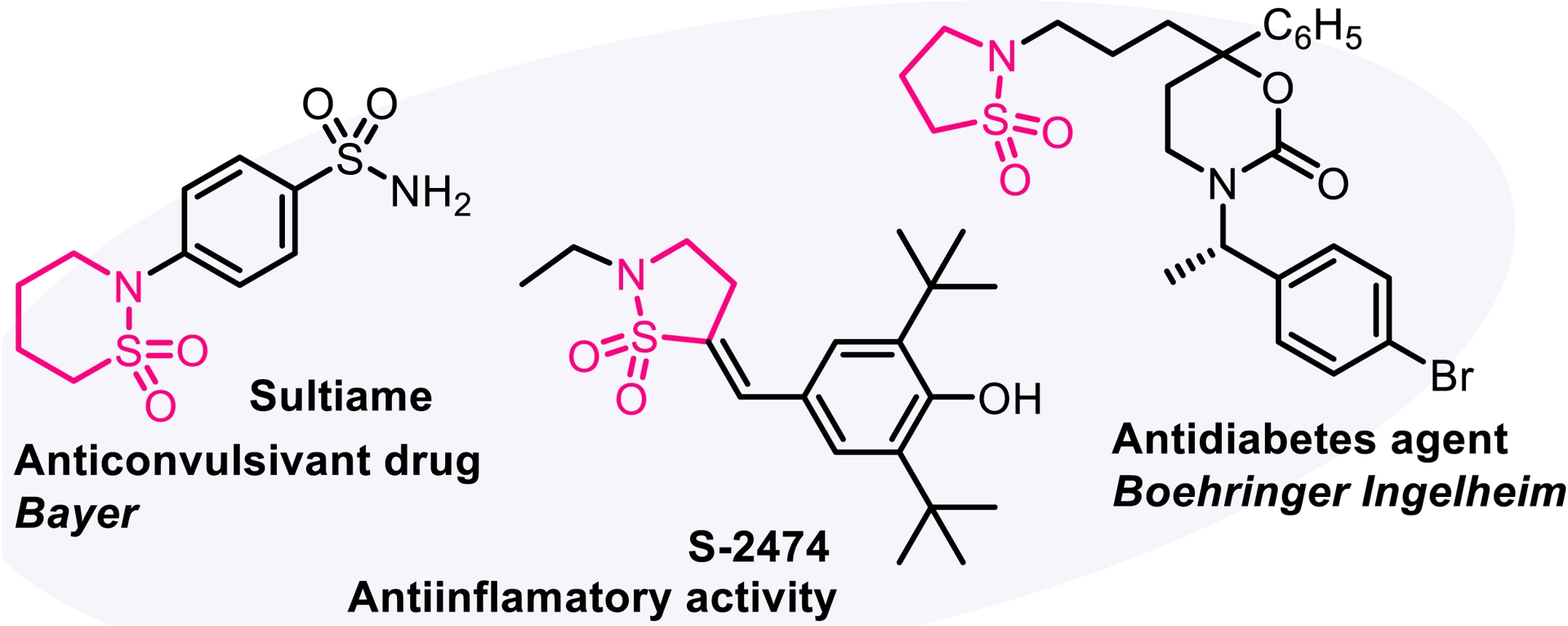
Synthesis of tri- and difluorinated γ-sultams.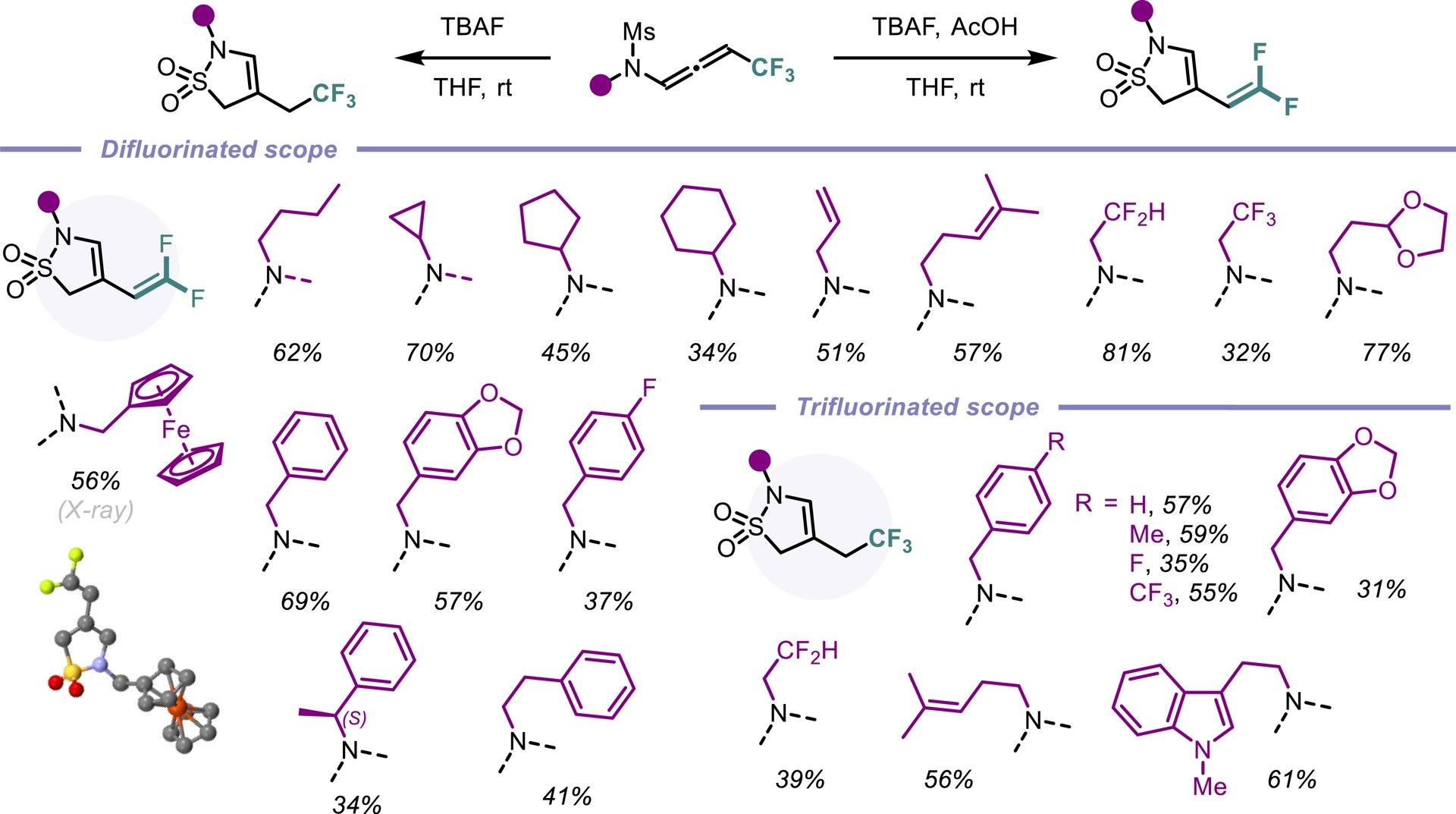
We next considered a chemoselective, metal-free approach for the 1,2- and 2,3-semireduction of CF3-substituted N-allenamides. The enamide moiety in these compounds was efficiently and regioselectively reduced using a combination of triethylsilane (Et3SiH) and boron trifluoride etherate (BF3⋅OEt2), affording the desired products with good stereoselectivity. Subsequent 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)-mediated isomerization of the resulting allyl amide proceeded cleanly, furnishing the E-configured enamide exclusively (Scheme 11) [51].
Semireduction and isomerization of $\text {CF}_3^-$ N-allenamides.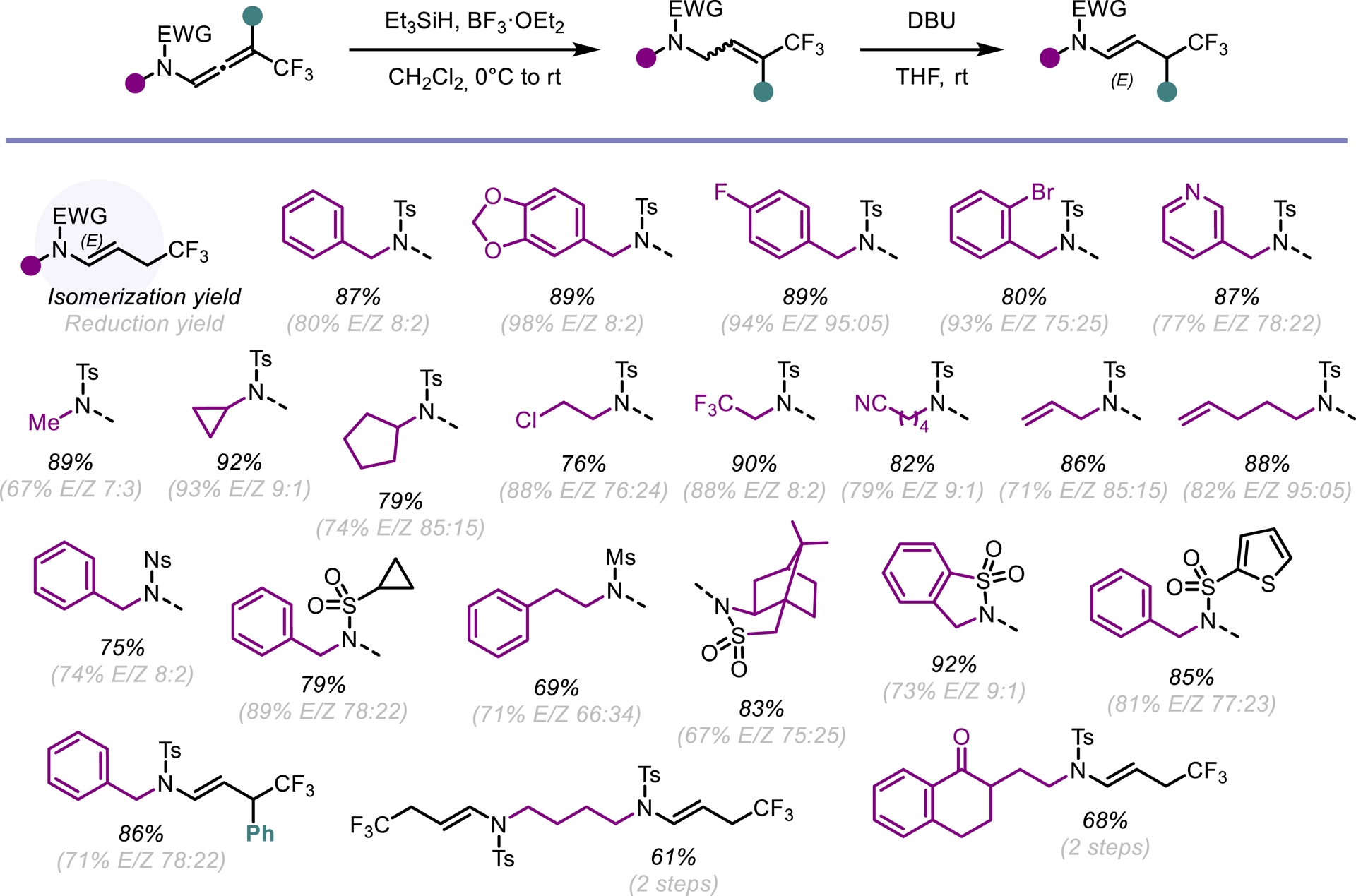
Among diverse organic azide compounds, vinyl azides represent a remarkable synthetic scaffold with their ability to act as an electrophile, an enamine-type nucleophile or a radical acceptor [52]. Given the unsaturated nature of N-allenamides, we envisioned that these compounds could be the perfect candidates to obtain vinyl azides via hydroazidations. We initiated our study by exploiting the reactivity of activated N-allenamides to achieve direct hydrofunctionalization at the central sp-hybridized carbon. Employing a combination of trimethylsilyl azide (TMSN3) and TBAF, we achieved a smooth hydroazidation of both trifluorinated and ester-substituted N-allenamides. This transformation yielded vinyl azides with complete regio- and stereoselectivity (Scheme 12). In contrast, when trifluorinated N-allenamides were activated with trifluoroacetic acid (TFA) in the presence of TMSN3, hydroazidation occurred selectively at the distal double bond. This enabled the site-specific installation of an azido group at the γ-position. While vinyl azides have found extensive utility in organic synthesis, allyl azides have often been overlooked due to their exposure to the Winstein rearrangement, typically leading to a mixture of isomers through allylic azide equilibrium [53]. In our system, the proximity of a heteroatom appears to suppress this equilibrium, enabling the exclusive formation of allyl azides featuring a conjugated system. Furthermore, treatment of these allyl azides with DBU induced a sigmatropic shift of the azido group, followed by double-bond isomerization. This process afforded vinyl azides with the azido moiety at the α-site with complete regio- and stereocontrol (Scheme 13) [54].
β-Hydroazidation of activated N-allenamides.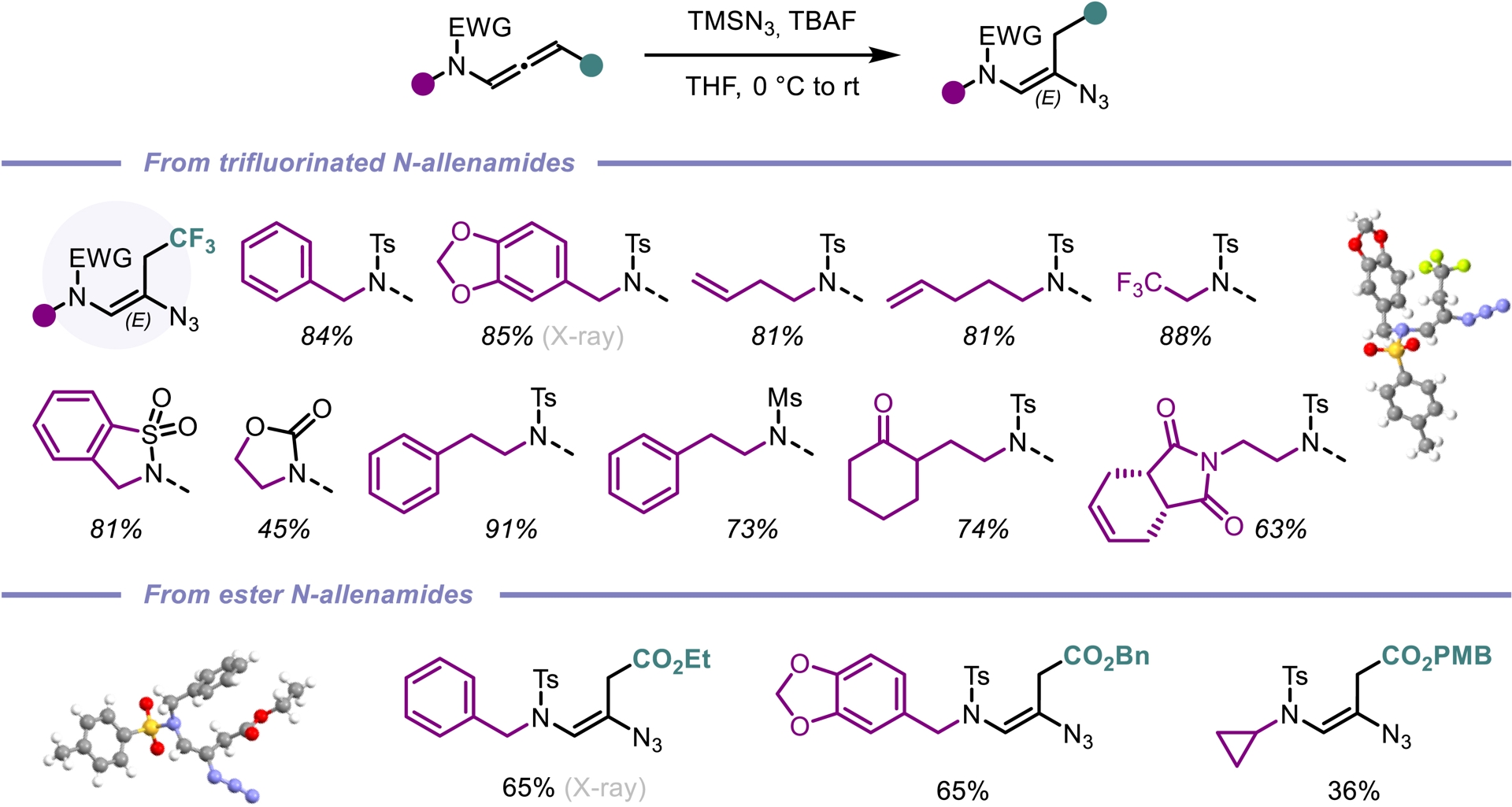
γ-Hydroazidation of activated N-allenamides and base-promoted azide shift.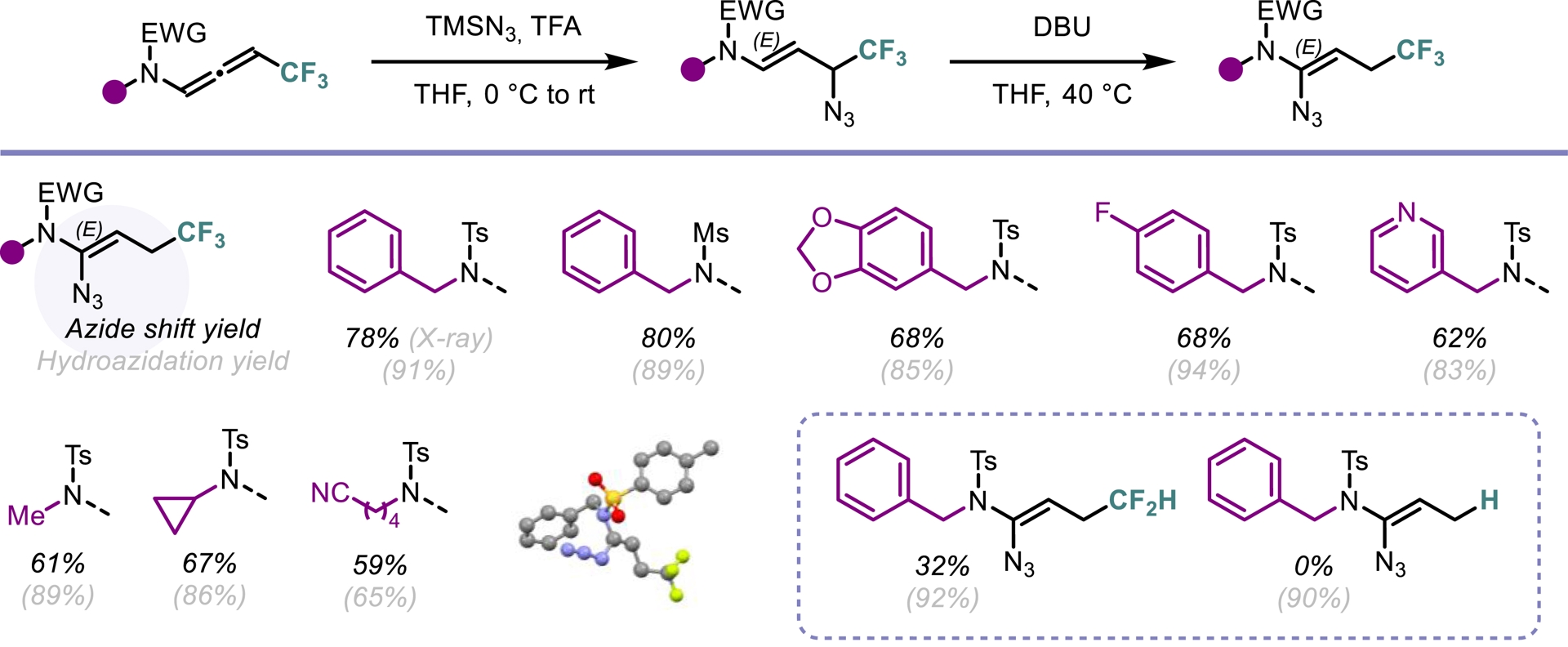
N-Allenamides bearing an electron-withdrawing group on the nitrogen atom have emerged as readily accessible and versatile nitrogen-containing synthons [1, 2, 3, 4, 5]. While their thermal reactivity—driven by electronic bias in the ground state (S0)—is well established [55, 56, 57], their behavior and reactivity in the triplet excited state (T1) remain largely unexplored. We were puzzled by the fact that the photochemical reactivity of N-sulfonyl allenamides remained unexplored (Figure 5) [58, 59]. We hypothesized that the presence of the sulfonyl group could be strategically leveraged to control the fate of the vinyl radical intermediate, potentially unlocking new reactivity pathways. Our investigations revealed that the direct addition of H-phosphine oxide to a CF3-substituted N-sulfonyl allenamide under visible-light irradiation led to the formation of the corresponding α-phosphorylated β-sulfonyl enamine. This transformation proved to be applicable to a broad range of substrates and demonstrated notable sustainability (Scheme 14). Remarkably, the reaction could be performed under natural sunlight over the course of two days, affording the desired phosphorylated enamine in 77% yield. Encouraged by our findings and the visible-light-induced N-to-C [1,3]-sulfonyl shift, we questioned whether this transposition could be extended beyond CF3-substituted N-allenamides. When terminal N-allenamides were employed in the presence of H-phosphine oxides, a divergent reactivity was observed, leading to the formation of structurally distinct hydrophosphorylated products. In this case, we propose that an intermolecular hydrogen atom transfer (HAT) occurs, followed by the addition of a phosphinoyl radical [P(O)⋅] to the terminal alkene moiety. This sequence furnishes γ-phosphorylated β-sulfonyl enamines. Notably, for terminal allenamides, functionalized side chains could be used and underwent a [1,3]-Ts shift, further expanding the structural diversity accessible through this approach. Additionally, terminal N-sulfonyl allenamides bearing various aryl and heteroaryl sulfonyl groups on the nitrogen atom were also effective substrates for this N-to-C [1,3]-sulfonyl shift reaction. This simple, efficient, and atom-economical process exhibits a broad substrate scope, excellent functional compatibility, and more importantly complete regio- and stereo-selectivity (Scheme 15) [60]. In conclusion, this account highlights our recent efforts toward the synthesis of activated N-allenamides and, more importantly, showcases their remarkable versatility as reactive intermediates. Under basic conditions, difluorinated ene-ynamides were efficiently generated. Nucleophilic additions to the difluorinated alkenyl moiety afforded highly functionalized, tailor-made dienes, while reactions with primary amines led to the formation of fluorinated pyrroles. Furthermore, the electron-withdrawing group on the nitrogen atom was strategically exploited to access potentially bioactive gem-difluorinated γ-sultams. Regio- and stereoselective reductions, as well as hydroazidations, delivered valuable fluorinated enamides, allylamides, and vinyl azides. Finally, the discovery of the first N-to-C tosyl shift in N-allenamides under photocatalyst-free conditions underscores the untapped potential of these compounds to unlock novel reactivity patterns and generate original phosphorylated structures.
The reactivity of excited N-allenamides: a chemical challenge.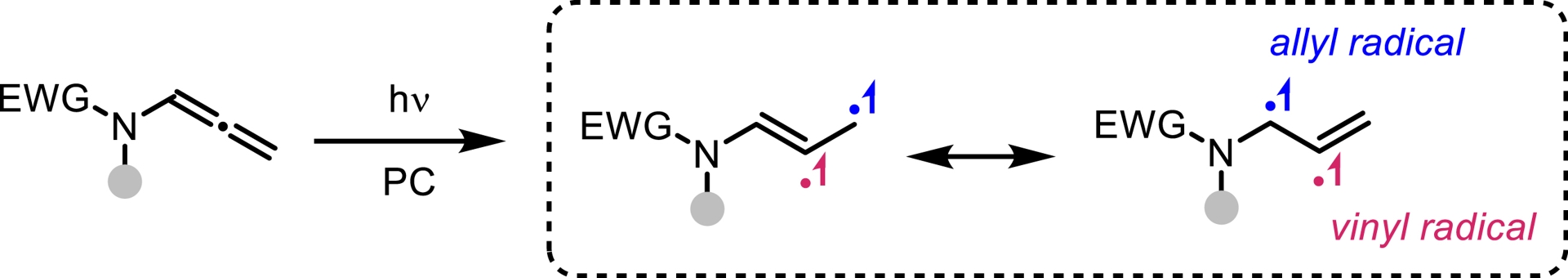
Light-induced synthesis of α-phosphorylated β-sulfonyl enamines.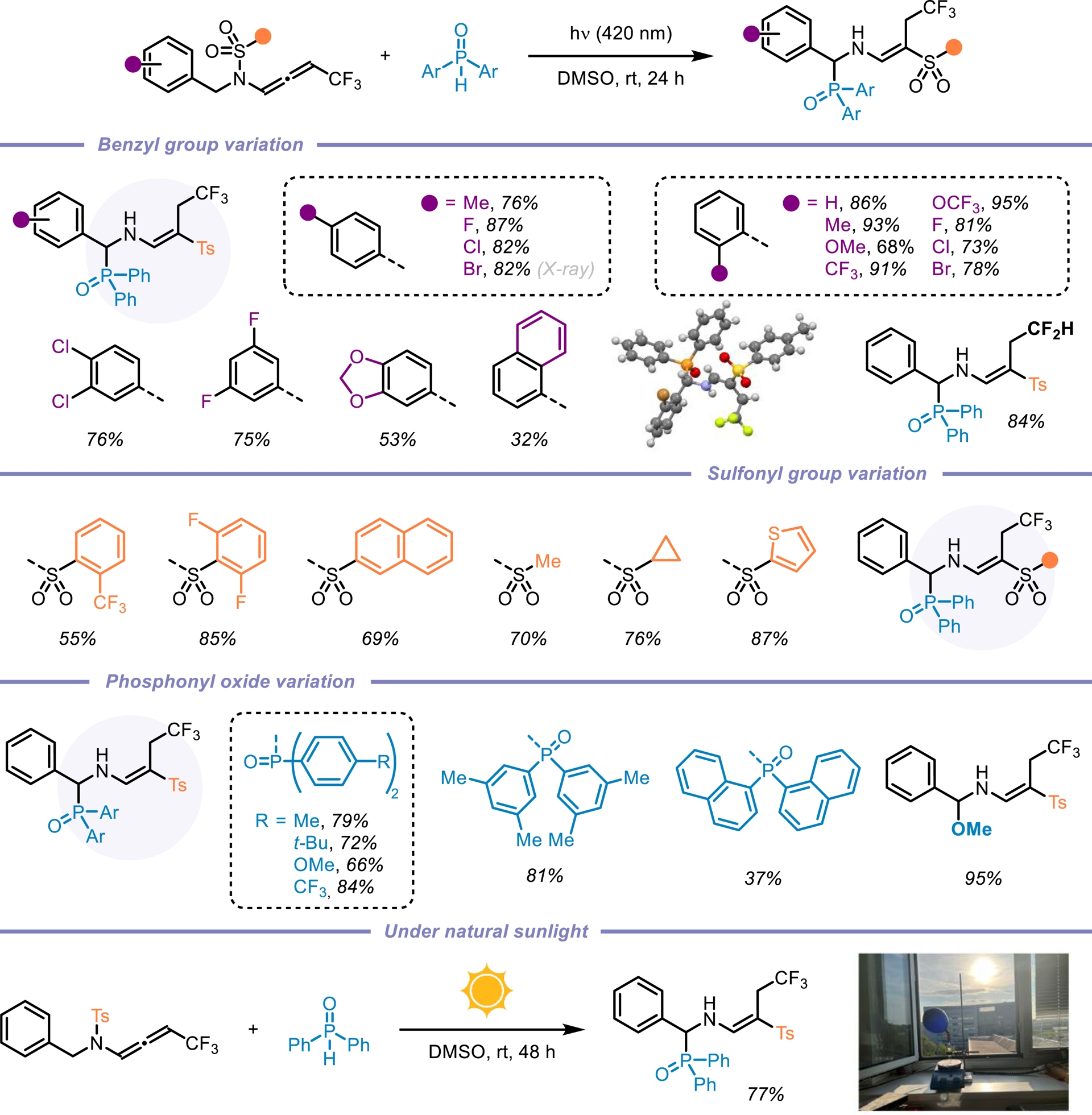
Light-induced synthesis of γ-phosphorylated β-sulfonyl enamines.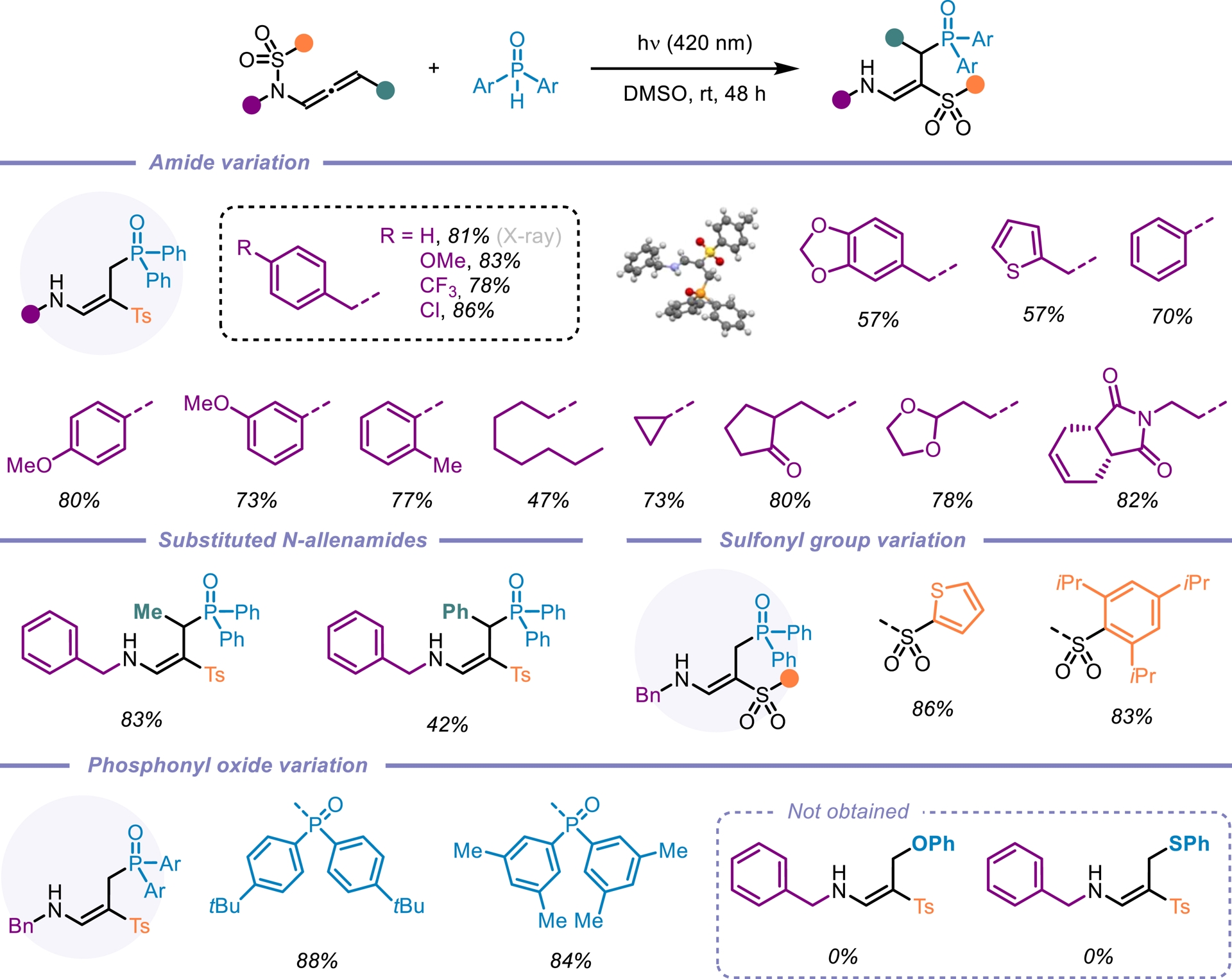
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.