

1 Introduction
L’utilisation des hétéropolyacides de type Keggin (HPA) de formule générale Hn[XM12O40], où X = P ou Si et M = Mo ou W ne cesse de s’accroître ces dernières années tant en catalyse d’oxydation [1–9], qu’en catalyse acide [10–13], aussi bien en milieu homogène qu’en milieu hétérogène. Leurs performances catalytiques sont essentiellement liées à leur caractère bifonctionnel (redox et acido-basique). Cependant, les inconvénients majeurs des hétéropolyacides phosphomolybdiques sont leur faible surface spécifique (≈ 10 m2/g) et leur mauvaise stabilité thermique.
Il a été rapporté que l’utilisation d’un support permettait d’améliorer les propriétés catalytique et thermique des hétéropolyacides. Parmi les différents supports utilisés, tels que SiO2 [14], charbon actif [15], TiO2 [16], polymères [17], MCM-41 [18–20], SBA-3 [21], SM-MCF [22], SBA-15 [23], HMS [6,24–26] et CMI-1 [27], les matériaux silicates à mésoporosité contrôlée ont été trouvés particulièrement attractifs. Ce sont des tamis moléculaires de grande surface spécifique (∼ 1000 m2/g), de volume poreux élevé (jusqu’à 1,8 cm3/g) et avec des pores dont la taille est comprise entre 2 et 10 nm. En plus, ils présentent des stabilités hydrothermale et thermique importantes. Il a été montré que les propriétés des HPAs supportés sont liées à la nature du support et à la masse de la phase active. Ainsi, les supports basiques avec de faibles charges d’espèces actives conduisent à la décomposition de l’hétéropolyacide, contrairement aux supports acides ou neutres. Pour éviter cette décomposition, il a été conseillé d’utiliser des pourcentages élevés en masse active (20–50 %) [18,19,23,24].
Dans ce travail, le choix s’est porté sur l’utilisation de matériaux silicates mésoporeux de type Hexagonal Mesoporous Silica (HMS), Chimie des matériaux inorganiques (CMI-1) et Santa Barbara no 15 (SBA-15) pour supporter l’hétéropolyacide, H4PMo11VO40. La méthode d’imprégnation sèche a été appliquée en utilisant 30 % en poids d’acide. Les matériaux ont été caractérisés par différentes techniques et testés dans l’oxydation du propène à 350 °C.
2 Partie expérimentale
2.1 Préparation des matériaux
Les matériaux HMS, CMI-1 et SBA-15 ont été préparés selon les procédés rapportés par Tanev et al. [28,29], Léonard et al. [30–32] et Stucky et al. [33,34], respectivement. L’hétéropolyacide H4PMo11VO40 noté (PMo11V) a été synthétisé selon la méthode classique de Tsigdinos et Hallada [35]. Trente pour cent de PMo11V/HMS, 30 % de PMo11V/CMI-1 et 30 % de PMo11V/SBA-15 ont été préparés par imprégnation sèche : un volume d’une solution aqueuse de H4PMo11VO40, 15H2O (pH 0,75) est versé goutte à goutte sous agitation sur 700 mg de support. Après séchage à 50 °C pendant 20 heures sous air, le solide est broyé dans un mortier. Le volume de la solution d’acide utilisé est idéalement égal au volume poreux du support (mesuré par physisorption d’azote) qui est de 1,45, 0,92 et 1,2 cm3/g pour HMS, CMI-1 et SBA-15, respectivement.
2.2 Caractérisation physico-chimique
Les compositions chimiques et pourcentages en poids de P, Mo, V et Si dans les matériaux ont été déterminés sur un spectrophotomètre d’émission atomique (AES) de type « ICP Thermo Jarrel Ash IRIS ADVANTAGE » à lecture radiale, équipé d’un détecteur CID couvrant des longueurs d’ondes de 175 à 800 nm.
Les mesures de surface spécifique et de porosité ont été effectuées par physisorption d’azote en utilisant un appareil de type Tristar 3000 de Micromeritics, après dégazage sous un vide de 5,33–6,67 Pa pendant 15 heures, à 350 °C pour les supports et à 130 °C pour les autres solides.
Les diffractogrammes des RX ont été réalisés dans la zone de 2θ variant entre 0,5° et 10° en utilisant un diffractomètre Panalytical X’pert pro et dans la gamme variant entre 10° et 70° sur un diffractomètre Siemens D5000, en utilisant dans les deux cas la raie Cu Kα (λ = 1,5418 Å) à 40 kV et 40 mA. L’indexation des raies de diffraction et l’identification des phases ont été réalisées en utilisant un logiciel DIFFRACTAT comprenant une base de données des fichiers ICDD-JCPDS.
Les spectres FT−IR et DRIFT ont été enregistrés à température ambiante au moyen d’un spectromètre infrarouge Equinox 55 (Brücker) à transformée de Fourier, dans la région 4400–370 cm−1, avec une résolution de 4 cm−1. Le rapport signal/bruit a été optimisé par accumulation de 100 interférogrammes par spectre dans le cas des analyses effectuées en mode transmission en utilisant 30–80 % en poids d’échantillon, mélangés avec du KBr sec et pressés sous forme de pastilles à 4 tonnes/cm2 et 200 interférogrammes par spectre dans le cas du DRIFT. Pour ce dernier, l’échantillon dilué avec du KBr (1:100) est placé dans une cellule Spectratech, opérant sous air afin de diminuer le risque de réduction.
L’analyse par MEB a été effectuée à l’aide d’un microscope LEO 983 GEMINI, équipé d’une source d’émission d’électrons fonctionnant avec une tension d’accélération de 1 kV.
Les analyses termogravimétrique et thermique différentielle (TG–ATD) ont été réalisées avec un appareil Mettler Toledo TGA/SDTA 851. Des masses d’environ 2 à 50 mg d’échantillons, mises dans des creusets en alumine, sont soumises à des traitements de 25 à 500, 600 ou 1000 °C, pour HPA, HPA/support ou support seul, respectivement, avec une vitesse de chauffe de 10 °C/min, sous un flux d’air horizontal de 100 mL/min.
Les analyses XPS ont été exécutées sur un spectromètre Kratos Axis Ultra (de la firme Kratos Analytical, Manchester, Royaume-Uni). Les mesures ont été réalisées grâce à une source de rayons X monochromatique issue de la raie Kα de l’aluminium (le courant d’émission de la source est fixé à 10 mA et 15 kV). Les échantillons pulvérulents ont été pressés dans des cupules en acier inoxydable de 4 mm de diamètre et de 0,5 mm de profondeur et introduits la veille de l’analyse dans la chambre de prétraitement du spectromètre à température ambiante. La pression dans la chambre de l’analyse est de l’ordre de 10−6 Pa. L’angle entre la normale à la surface de l’échantillon et l’axe du détecteur est de 0°. Le mode hybride du grossissement de l’objectif a été utilisé avec une ouverture de fente qui donne une région analysée de 700 μm × 300 μm. L’énergie de passage de l’analyseur hémisphérique est de 40 eV. La charge développée en surface est compensée par un canon à électrons couplé à une lentille magnétique. Dans ces conditions, la résolution d’énergie donne une largeur à mi-hauteur (FWHM) du pic de l’Ag 3d5/2 à environ 1,0 eV. La stabilisation de la charge est réalisée en utilisant l’Axe Kratos de l’appareil. La séquence suivante de spectres a été enregistrée : C 1s, O 1s, Si 2p, P 2p, Mo 3d, V 2p et C 1s une seconde fois pour vérifier l’effet de charge en fonction du temps et l’absence de dégradation de l’échantillon durant l’analyse. Les énergies de liaison sont calculées par rapport à la référence interne C−(C,H) du pic C 1s fixé à 284,8 eV. L’énergie de passage au niveau de l’analyseur est de 160 eV. Les spectres ont été décomposés avec le programme CasaXPS (Logiciel Casa Ltd., Royaume-Uni) avec un rapport Gaussian/Lorentzian (70/30 %) et après soustraction de la ligne de base linéaire. Les fractions molaires ont été calculées en utilisant des pics de surfaces normalisés sur la base des paramètres d’acquisition, des facteurs de sensibilité fournis par le fabricant et de la fonction de transmission.
2.3 Activité catalytique
L’oxydation du propène a été réalisée à pression atmosphérique dans un domaine de températures de 200 à 400 °C, en utilisant un microréacteur tubulaire en quartz sous forme de U à lit fixe. La température de la réaction a été réglée avec un régulateur PID couplé à un thermocouple coaxial, en contact avec le lit catalytique. Deux cent dix, 90 ou 300 mg de support seul, HPA à l’état massique ou HPA supporté, respectivement ont été utilisés (Ø comprise entre 200 et 315 μm). Au-dessus du catalyseur ont été introduits des grains de quartz (Ø > 315 μm) en remplissant toute la section du réacteur. Le catalyseur a été chauffé jusqu’à la température de réaction (10 °C/min) en présence du mélange réactionnel composé de 10 % C3H6, 20 % O2 et 70 % He, dont le rapport en volume est de 1/2/7. Le débit total est de 30 mL/min. Les analyses des réactifs et produits ont été réalisées en ligne, en utilisant un chromatographe DELSI NERMAG équipé d’un FID pour les produits oxygénés. Les hydrocarbures, COx et O2 ont été analysés avec un chromatographe INTERSMAT IGC 12 M équipé d’un TCD et possédant deux colonnes Haysep Q (2 m) et molecular sieve 5A (2 m) montées en parallèle à 80 °C.
3 Résultats expérimentaux
3.1 Analyse chimique
Le Tableau 1 représente les résultats de l’analyse chimique des différents matériaux. Avant et après imprégnation de l’hétéropolyacide sur les supports HMS, CMI-1 et SBA-15, les nombres d’atomes de phosphore (0,98–1,01) et de vanadium (1,00–1,02) calculés en considérant 11 atomes de molybdène par unité de Keggin sont en très bon accord avec les valeurs théoriques.
Composition chimique à partir de l’ICP de PMo11V, HMS, CMI-1, SBA-15, PMo11V/HMS, PMo11V/CMI-1 et PMo11V/SBA-15.
| Matériaux | Quantité | Pa | Moa | Va | P/Si | Mo/Si | V/Si | |||
| Si (%) | P (%) | Mo (%) | V (%) | |||||||
| PMo11V | − | 1,44 | 49,34 | 2,39 | 0,99 | 11,00 | 1,00 | − | − | − |
| HMS | 44,91 | − | − | − | − | − | − | − | − | − |
| CMI-1 | 41,62 | − | − | − | − | − | − | − | − | − |
| SBA-15 | 41,25 | − | − | − | − | − | − | − | − | − |
| PMo11V/HMS | 30,27 | 0,39 | 13,61 | 0,65 | 0,98 | 11,00 | 1,00 | 0,012 | 0,132 | 0,012 |
| PMo11V/CMI-1 | 28,66 | 0,40 | 13,63 | 0,66 | 1,00 | 11,00 | 1,00 | 0,013 | 0,139 | 0,013 |
| PMo11V/SBA-15 | 27,30 | 0,46 | 15,54 | 0,76 | 1,01 | 11,00 | 1,02 | 0,015 | 0,167 | 0,015 |
a Nombre d’atomes par unité de Keggin [PMo11V]4−.
3.2 Caractérisation texturale
Les isothermes d’adsorption–désorption d’azote correspondant aux différents matériaux mésoporeux, HMS, CMI-1 et SBA-15 sont de type IV (Fig. 1) (cf. IUPAC), avec des boucles d’hystérésis de type H1 caractérisées par des paliers de saturation. Il a été rapporté que la boucle d’hystérésis H1 est caractéristique des matériaux mésoporeux avec une distribution étroite de pores [30,32,36–39].
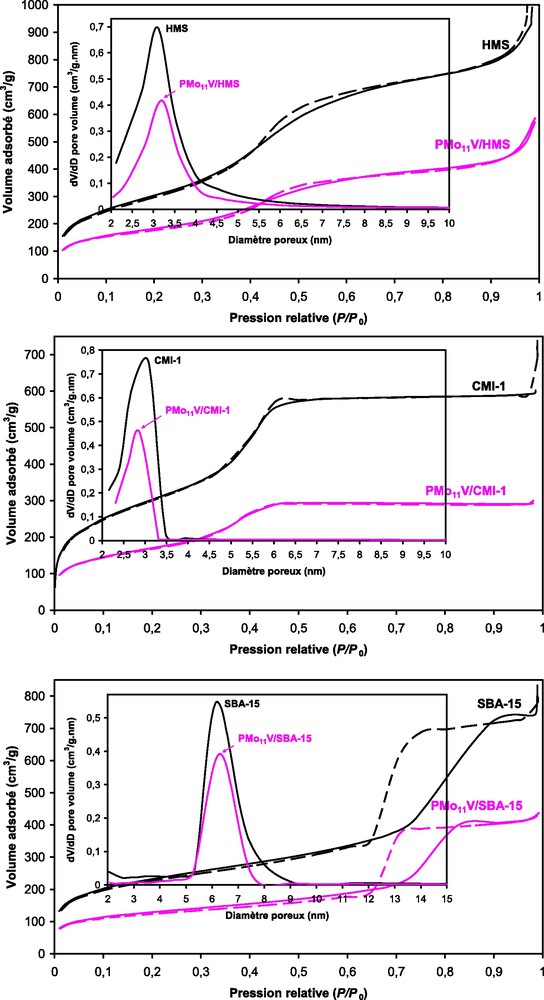
Isothermes d’adsorption–désorption d’azote et distributions de taille de pores déterminées par BJH des matériaux mésoporeux purs et supportés de PMo11V.
Sur ces isothermes, on distingue trois régions. Selon la théorie BET, la première région située aux faibles pressions relatives correspond à la formation d’une monocouche. La seconde région correspondant à la condensation capillaire, est observée dans les cas de la HMS et la CMI-1 au-delà d’une pression relative de l’ordre de 0,4 et dans le cas de la SBA-15 à une pression relative plus élevée (∼ 0,65). Cette condensation renseigne sur la taille des pores. La troisième région située au-delà des pressions relatives de 0,52 pour la CMI-1 et 0,9 pour la SBA-15, correspond à un plateau de saturation. Ce plateau suggère pour ces deux matériaux que la macroporosité interparticulaire est faible. Dans le cas de la HMS, une croissance asymptotique est observée à P/P0 > 0,85. Cette croissance suggère une macroporosité de texture interparticulaire significative [22].
L’ajout de l’hétéropolyacide, PMo11V, au support conduit à des isothermes d’adsorption–désorption d’azote (Fig. 1) similaires à celles des matériaux purs avec cependant des inflexions moins importantes indiquant une diminution de la mésoporosité des supports, notamment dans le cas de la HMS et la CMI-1 montrant que leurs mésopores se retrouvent ainsi fermés à un bout, alors que la réminiscence de la boucle d’hystérèse dans le cas de la SBA-15 montre que les mésopores restent ouverts aux deux extrémités.
Le Tableau 2 montre les différents résultats de caractérisation physique des solides. La HMS et la CMI-1 possèdent des surfaces spécifiques de 1129 et 1114 m2/g, respectivement, supérieures à celle de la SBA-15 (792 m2/g) et des diamètres de pore similaires 3,10–3,08 nm, mais inférieurs d’un facteur de 2 à celui de la SBA-15 (6,17 nm). Cette dernière possède des parois environ deux fois plus larges que celles des deux autres matériaux, CMI-1 et HMS (4,80 contre 2,53 et 2,36 nm, respectivement). Le volume poreux ne suit pas la même tendance. Ainsi, HMS et SBA-15 ont des volumes poreux du même ordre de grandeur (1,45 et 1,20 cm3/g, respectivement), supérieurs à celui de la CMI-1 (0,92 cm3/g). Ces différences physiques entre les trois supports sont probablement liées à la morphologie du matériau comme le montrent les micrographies MEB (Fig. 3). Ces résultats montrent que les matériaux synthétisés sont bien des matériaux mésoporeux qui diffèrent par leurs propriétés physiques.
Caractéristiques physiques des solides PMo11V, HMS, CMI-1, SBA-15, PMo11V/HMS, PMo11V/CMI-1 et PMo11V/SBA-15.
| Matériaux | SBET (m2/g) | ∅BJHc (nm) | Volume poreuxd (cm3/g) | d(1,0,0) (nm) | a0e (nm) | WTtf (nm) |
| V1 | 6a | 5,51 | 0,01 | − | − | − |
| HMS | 1129b | 3,10 | 1,45 | 4,73 | 5,46 | 2,36 |
| CMI-1 | 1114b | 3,08 | 0,92 | 4,86 | 5,61 | 2,53 |
| SBA-15 | 792b | 6,17 | 1,20 | 9,50 | 10,97 | 4,80 |
| PMo11V/HMS | 650a | 3,20 | 0,84 | 4,65 | 5,37 | 2,17 |
| PMo11V/CMI-1 | 610a | 2,88 | 0,45 | 4,82 | 5,57 | 2,69 |
| PMo11V/SBA-15 | 437a | 6,31 | 0,65 | 9,85 | 11,37 | 5,06 |
a Surface BET mesurée après dégazage à 130 °C.
b Surface BET mesurée après dégazage à 350 °C.
c Diamètre poreux moyen déterminé par la méthode B.J.H (volume poreux dV/dD, modèle des pores cylindriques).
d Volume poreux déterminé à partir de l’isotherme d’adsorption–désorption d’azote pour P/P0 = 0,98–0,99.
e Paramètre de maille a0 = 2d100/√3.
f Épaisseur moyenne des parois WTt = a0 – ∅BJH.

Micrographies MEB des matériaux mésoporeux (a) HMS, (b) PMo11V/HMS, (c) CMI-1, (d) PMo11V/CMI-1, (e) SBA-15 et (f) PMo11V/SBA-15.
Après imprégnation de 30 % en poids de H4PMo11VO40, la surface BET (SBET) et le volume poreux (Vp) des trois supports, diminuent d’un facteur de l’ordre de 2. Ainsi, ils passent respectivement pour la HMS, de 1129 à 650 m2/g et de 1,45 à 0,84 cm3/g, pour la CMI-1, de 1114 à 610 m2/g et de 0,92 à 0,45 cm3/g et pour la SBA-15, de 792 à 437 m2/g et de 1,2 à 0,65 cm3/g. La diminution de la surface externe et celle du volume poreux dans les systèmes HPAs supportés sur MCM-41 [18,19,40], SBA-3 [21] et HMS [24,25,41] ont déjà été rapportées. Cela a été attribué au colmatage des mésopores du support par les hétéropolyanions ou à l’effondrement partiel des parois du support. La diminution de ces paramètres peut être également expliquée par la pénétration des molécules du HPA dans les mésopores des supports. En effet, cette pénétration pourrait se produire en raison du diamètre de l’unité de Keggin qui est d’environ 12 Å comparé à celui des mésopores qui est de 31, 30,8 et 61,7 Å pour la HMS, CMI-1 et SBA-15, respectivement. Cependant, dans le cas de ce travail, le fait que le volume poreux diminue, alors que le diamètre des pores semble le même après introduction du HPA (3,10-3,20, 3,08-2,88 et 6,17-6,31 nm en présence de HMS, CMI-1 et SBA-15, respectivement) suggèrerait sur la base de l’hypothèse proposée par Navez et al. [42] que les espèces actives ne pénètrent pas à l’intérieur des pores mais restent plutôt à la surface du support.
Le Tableau 2 montre également que les caractéristiques cristallographiques du matériau SBA-15 se distinguent de celles des deux autres matériaux avec des paramètres a0 de 10,97 nm et d100 de 9,50 nm contre, respectivement 5,46 nm et 4,73 nm pour la HMS et 5,61 nm et 4,86 nm pour la CMI-1. La variation de ces deux paramètres est plus marquée après imprégnation de l’hétéropolyacide sur la SBA-15 avec une augmentation de a0 de 10,97 à 11,37 nm et de 9,50 à 9,85 nm pour d100.
3.3 Diffraction des Rayons X (DRX)
La Fig. 2 montre les diffractogrammes RX des différents systèmes aux bas (2θ : 0,5−10°) et hauts angles (2θ : 10−70°). Contrairement au diffractogramme RX de la HMS montrant un seul pic à 2θ = 1,9° correspondant au plan (1 0 0), ceux de la CMI-1 et la SBA-15 (Fig. 2b et c) montrent trois pics relativement bien résolus dans la zone d’angles de 2θ : 1 − 4°, qui peuvent être indexés comme suit (1 0 0), (1 1 0) et (2 0 0). Le pic intense (1 0 0) attribué à la réflexion primaire confirme l’homogénéité de la taille de pores. Il est situé à 2θ = 0,93° pour la SBA-15, et 2θ = 1,8° pour la CMI-1. La présence des pics de réflexions secondaires (1 1 0) à 2θ = 3,1° et (2 0 0) à 2θ = 3,6° sur le diffractogramme de la CMI-1 et à 2θ = 1,5° et 2θ = 1,8°, respectivement sur celui de la SBA-15, confirme la symétrie hexagonale (p6 mm) dans l’arrangement des canaux de ces deux matériaux [30–33].
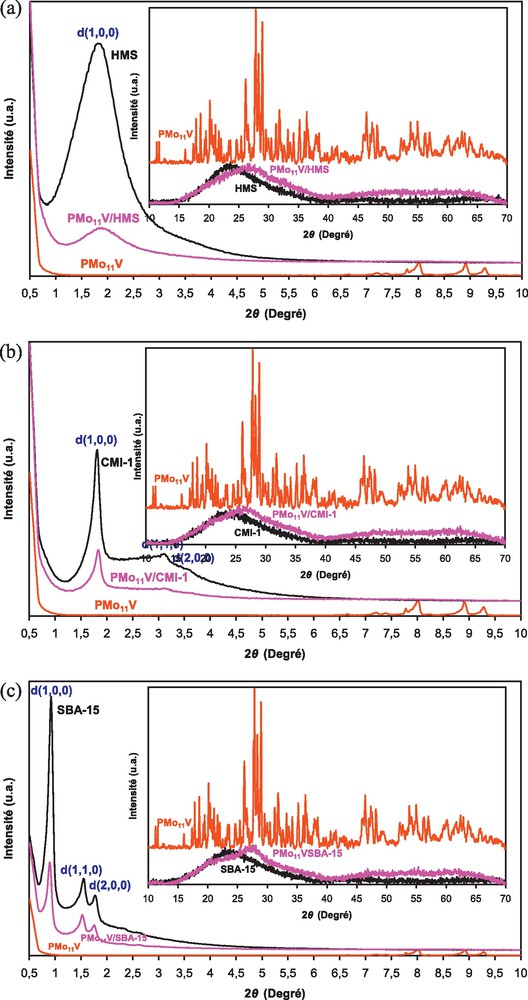
Diffractogrammes RX de (a) HMS, PMo11V–HMS, (b) CMI-1, PMo11V–CMI-1 et (d) SBA-15 et PMo11V–SBA-15, aux bas (2θ : 0,5–10°) et hauts angles (2θ : 10–70°).
Les diffractogrammes RX des systèmes, HPA/support, montrent les principales réflexions des matériaux HMS, CMI-1 ou SBA-15 mais avec un léger décalage et des intensités plus faibles. Cela suggère que leur structure mésoporeuse est préservée mais avec un effondrement partiel des parois dû à la présence de l’hétéropolyacide [6,18–20,27,38,40,43].
Aux hauts angles (Fig. 2), les diffractogrammes RX des trois supports présentent un large pic entre 2θ =15 et 40° assigné à la silice amorphe [14,41]. Contrairement aux bas angles, après imprégnation du PMo11V, le pic n’a pas diminué d’intensité mais s’est légèrement déplacé vers des angles plus élevés. Aucune phase cristalline du HPA n’est observée sur les diffractogrammes des systèmes, HPA/support. Cela suggère que les espèces hétéropolyanioniques sont finement dispersées à la surface du support [6,18,22,24,27,41,44,45].
3.4 Caractérisation morphologique par microscopie électronique à balayage (MEB)
Sur les micrographies MEB représentées sur la Fig. 3, la HMS se présente sous forme de chou-fleur uniforme formée d’agrégats de 0,5 à 50 μm (Fig. 3a). L’observation des particules des matrices CMI-1 (Fig. 3b) montre que ce matériau possède des particules de morphologies différentes (vers, gyroïdes et toroïdes) avec une taille de l’ordre de 5 μm [31,32]. Le matériau SBA-15 présente une morphologie de vers enchevêtrés de l’ordre de quelques micromètres (Fig. 3e). Cette morphologie fibreuse correspond à celle décrite dans une étude spécifique concernant le contrôle de la morphologie pour les matériaux SBA-15 [46].
L’ajout de l’hétéropolyacide sur ces différents supports ne semble pas altérer leur morphologie. Aucune forme cristalline de PMo11V n’a été observée à leur surface confirmant ainsi la bonne dispersion des espèces hétéropolyanioniques. Des résultats similaires ont été obtenus dans les cas de PW12–HMS [24] et de PMo10V2–HMS [41].
3.5 Études spectroscopiques FT–IR et DRIFT
Les spectres FT−R de PMo11V, du matériau mésoporeux SBA-15 pur et supporté sont illustrés sur la Fig. 4. La structure de Keggin de l’hétéropolyacide, PMo11V est identifiée par la présence de bandes de vibration IR dans la région de faibles longueurs d’onde (1100–500 cm−1). Selon Rocchiccioli-Deltcheff et al. [47], les bandes situées à 1063, 962, 866, 785 et 596 cm−1 correspondent aux vibrations νas(P–Oa), νas(M–Od), νas(M–Ob–M), νas(M–Oc–M) (M = Mo or V)et δ(P–O), respectivement. Dans l’unité de Keggin, Oa est commun au tétraèdre central PO4 et à un groupement trimétallique M3O13, Ob assure la jonction entre deux groupements trimétalliques, Oc lie deux octaèdres MO6 à l’intérieur d’un même groupement trimétallique et Od, appelé oxygène terminal est lié au métal par une double liaison (MOd).
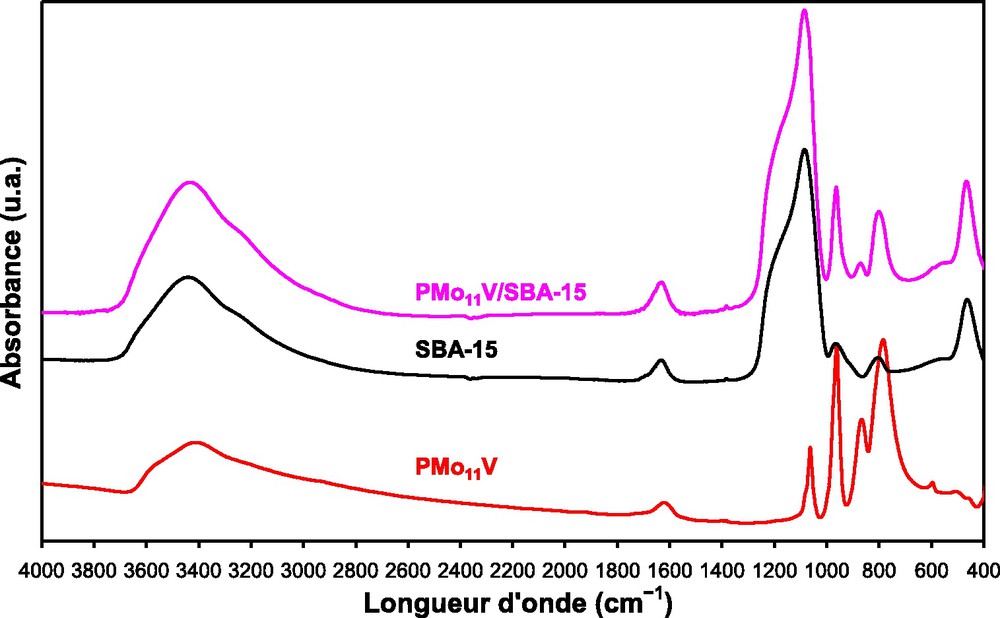
Spectres FT−IR de PMo11V, SBA-15 et PMo11V/SBA-15.
Le spectre IR de la SBA-15 exhibe des bandes de vibration dans la région 3700–400 cm−1, attribuées aux liaisons Si–O–Si, SiO4 et aux groupements OH [42,48–51].
Le spectre IR de l’hétéropolyacide, PMo11V supporté sur la SBA-15 montre que les bandes de vibration νas(M–Od) et νas(M–O–M) typiques de PMo11V situées à 962 et 866 + 785 cm−1, sont maintenues après imprégnation. Les autres bandes sont masquées par celles du support. Ces observations indiquent que la structure de Keggin a été préservée lors de l’imprégnation de l’acide sur le support [6,21,24–26,27,38]. Des résultats analogues ont été observés avec les supports HMS et CMI-1(figures non représentées).
Le spectre DRIFT de l’hétéropolyacide, PMo11V (Fig. 5) montre les bandes caractéristiques de la structure de Keggin dans la région 1100−700 cm−1. Les bandes d’absorption observées à 1063 et 962 cm−1 sont attribuées aux liaisons νas(P–O) et νas(M = O), respectivement et celles à 868 et 787 cm−1 à νas(M–O–M) (M = Mo, V) [52]. Le spectre DRIFT (Fig. 5) de la CMI-1 montre les bandes caractéristiques des groupements silanol, des vibrations Si–O, Si–O–Si et des vibrations H–O–H des molécules d’eau physisorbée [25,53–55]. Sur le spectre de PMo11V/CMI-1, les bandes de vibration correspondant à la structure de Keggin ont été entièrement masquées par celles du support dans la région 1800−400 cm−1, à l’exception de celles correspondant aux vibrations νas(M–Od) (962 cm−1) et νas(M–O–M) (868 + 787 cm−1). Comme dans le cas de l’analyse par spectroscopie FT–IR, cela indique que la structure de Keggin n’a pas été affectée par le support [6,27]. Cependant, après l’imprégnation de PMo11V, l’intensité de la bande à ∼ 3750 cm−1 du support diminue fortement et se déplace de 2 à 10 cm−1 vers les faibles longueurs d’onde. Cette diminution est provoquée par une forte interaction entre l’acide avec les groupements silanol conduisant ainsi à des espèces de surface (SiOH2+)(H3PMo11VO40−) [6,27,40,56,57]. Des résultats analogues ont été observés avec les supports HMS et SBA-15. La formation des espèces (SiOH2+)(H3PMo11VO40−) semble être indépendante de la nature du support mésoporeux.
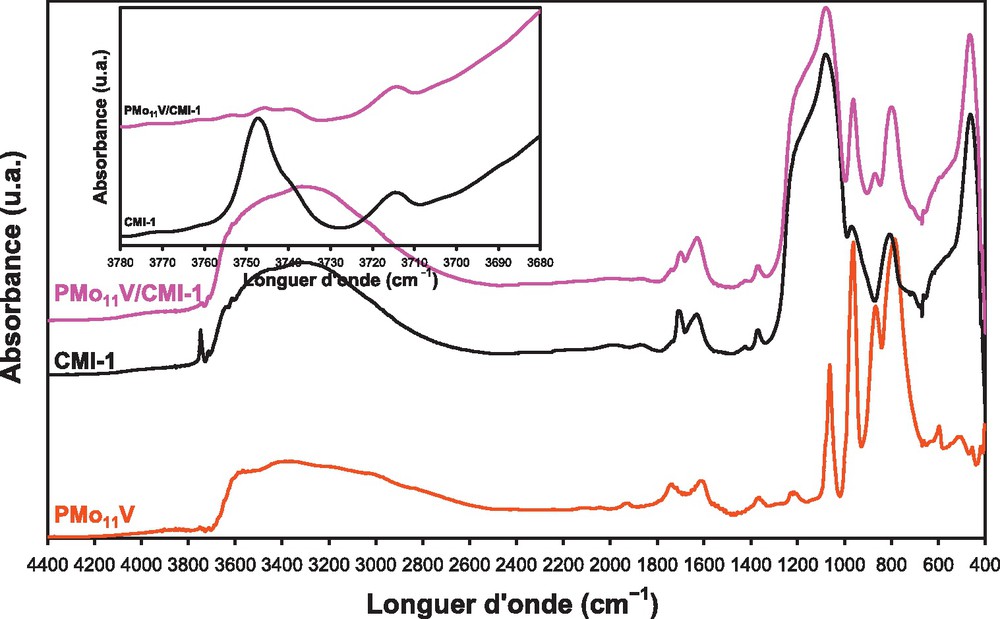
Spectres DRIFT de PMo11V, CMI-1 et PMo11V/CMI-1.
3.6 Analyse thermique (TG−ATD)
Selon les données de littérature [52,58], l’analyse thermogravimétrique du PMo11V (Fig. 6a) met en évidence la perte des molécules d’eau de cristallisation avant 200 °C et celle des molécules d’eau de constitution entre 200 et 450 °C. La première perte de masse représente les 15 molécules d’eau d’hydratation et la seconde les deux molécules d’eau associées aux quatre protons de l’acide. Ces pertes de poids correspondent aux pics endothermiques observés à 94, 123 et à 340 °C sur la courbe ATD. Le signal exothermique assigné à la décomposition du PMo11V en P2O5, V2O5 et MoO3 est observé vers 460 °C. L’analyse thermique des supports ne montre aucune perte de masse de 25 à 600 °C (figures non représentées).
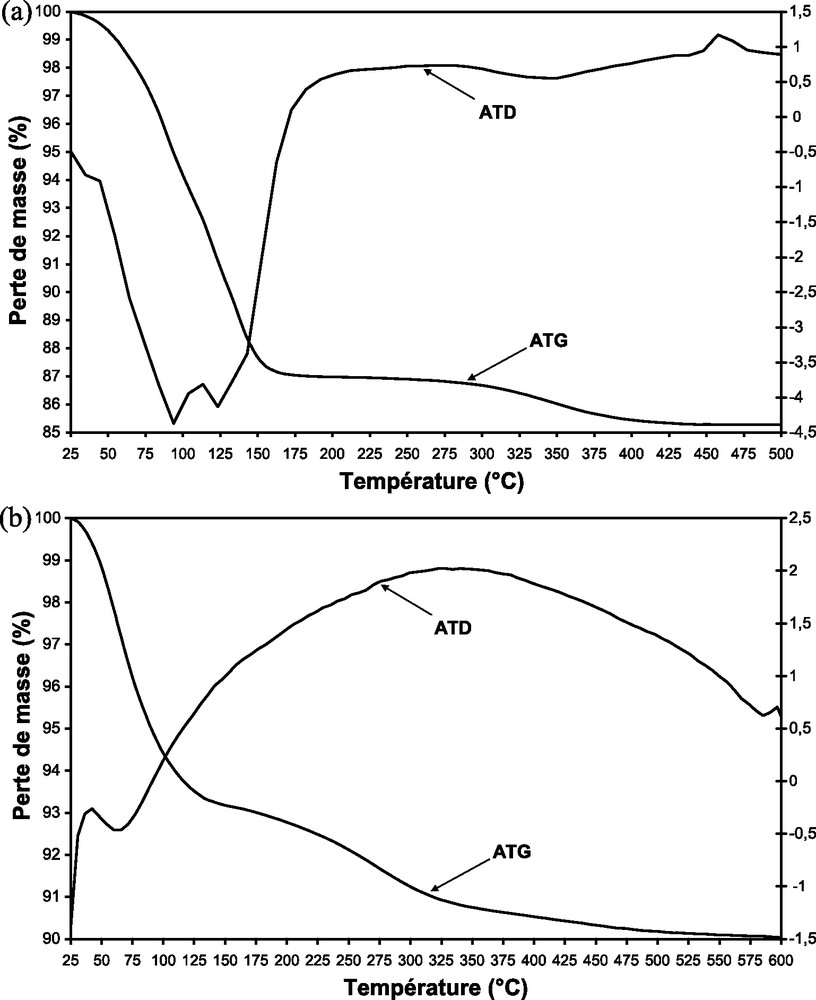
Courbes ATG et ATD de (a) PMo11V et (b) PMo11V/HMS.
La courbe TG du matériau supporté sur la HMS (Fig. 6b) est similaire à celle de l’acide non supporté mais avec une perte de masse progressive jusqu’à 600 °C, attribuée au départ des molécules d’eau de l’hétéropolyacide et probablement aux traces résiduelles du surfactant organique utilisé dans la synthèse de la HMS. Le pic exothermique assigné à la décomposition de l’hétéropolyacide est observé à une température plus élevée (600 °C) que celle correspondant à l’acide massique (460 °C). Ce résultat suggère que le support HMS stabilise le HPA suite à la formation des espèces de surface ( SiOH2+)(H3PMo11VO40−) comme constatée dans l’analyse DRIFT [6,27]. Ces espèces paraissent plus stables que la forme de l’acide libre (H4PMo11VO40). Des résultats analogues ont été obtenus avec les autres supports.
3.7 Spectroscopie de photoémission X (XPS)
Les Tableaux 3, 4 et 5 rassemblent les résultats XPS correspondant aux énergies de liaison (EL), fractions molaires de surface de chaque élément et aux rapports atomiques de surface rapportés à Si.
Énergies de liaison (eV) des différents éléments.
| PMo11V | HMS | CMI-1 | SBA-15 | PMo11V/HMS | PMo11V/CMI-1 | PMo11V/SBA-15 | |
| O 1sa | − | 533,0 | 533,2 | 533,1 | 533,0 | 533,1 | 533,0 |
| O 1sb | 531,0 | − | − | − | 530,9 | 531,0 | 531,0 |
| C 1s C-(C,H) | 284,8 | 284,8 | 284,8 | 284,8 | 284,8 | 284,8 | 284,8 |
| Si 2p | − | 103,7 | 103,9 | 103,8 | 103,9 | 103,9 | 103,7 |
| P 2p | 134,1 | − | − | − | 134,2 | 134,8 | 134,2 |
| Mo 3d | 233,3 | − | − | − | 233,3 | 233,4 | 233,4 |
| V 2p | 518,1 | − | − | − | 517,2 | 516,8 | 517,0 |
a Correspondant à l’oxygène du support.
b Correspondant à l’oxygène du PMo11V.
Fractions molaires (%) de surface des spectres individuels.
| PMo11V | HMS | CMI-1 | SBA-15 | PMo11V/HMS | PMo11V/CMI-1 | PMo11V/SBA-15 | |
| O 1s | 57,8 | 62,7 | 63,0 | 62,3 | 63,5 | 63,4 | 62,1 |
| C 1s | 18,8 | 1,7 | 2,5 | 4,3 | 2,8 | 2,8 | 5,1 |
| Si 2p | − | 35,6 | 34,5 | 33,4 | 32,2 | 32 | 30,2 |
| P 2p | 2,3 | − | − | − | 0,1 | 0,3 | 0,3 |
| Mo 3d | 20,0 | − | − | − | 1,3 | 1,3 | 2,1 |
| V 2p | 1,1 | − | − | − | 0,1 | 0,2 | 0,2 |
Rapports atomiques de surface (M/Si).
| HMS | CMI-1 | SBA-15 | PMo11V/HMS | PMo11V/CMI-1 | PMo11V/SBA-15 | |
| O/Si | 1,761 | 1,826 | 1,865 | 1,971 | 1,981 | 2,056 |
| C/Si | 0,048 | 0,049 | 0,129 | 0,086 | 0,087 | 0,169 |
| P/Si | − | − | − | 0,005 | 0,009 | 0,010 |
| Mo/Si | − | − | − | 0,041 | 0,041 | 0,066 |
| V/Si | − | − | − | 0,003 | 0,006 | 0,007 |
Les EL correspondant au silicium Si 2p des trois supports sont équivalentes (103,7–103,9 eV), après imprégnation de l’acide aucune variation significative de l’énergie de liaison n’est observée. Ces valeurs sont similaires à celles rapportées dans la littérature [25,37,50].
La décomposition du spectre de photoémission de la raie 1s de l’oxygène des échantillons imprégnés de PMo11V montre deux composantes, une principale centrée à 533,0–533,2 eV attribuée à l’oxygène des supports siliciques [25,37,50] et une autre de plus faible intensité située à 531,0 ± 0,1 eV, attribuée aux atomes d’oxygène de l’hétéropolyacide [16].
Les EL de 134,1 eV du niveau 2p3/2 du phosphore (V) [16] et de 233,3 ± 0,1 eV du niveau 3d5/2 du molybdène (VI) [16,26] de l’hétéropolyacide ne varient pratiquement pas après imprégnation sur les différents supports.
Comme il a été mentionné précédemment, la diminution de la valeur de EL de 518,1 eV correspondant au V 2p3/2 caractéristique de V(V) à 517,2 ± 0,2 eV et l’élargissement du pic V 2p3/2 après imprégnation de l’acide sont attribués à une photoréduction de l’échantillon suite à son exposition à la source de rayons X dans la chambre du spectromètre [26] et ne peuvent pas être discutés en termes de modification structurale du HPA.
Le Tableau 5 présente les rapports atomiques de surface M/Si (M : O, P, Mo et V) estimés à partir de l’analyse XPS.
L’échantillon PMo11V/SBA-15 conduit à des rapports M/Si (M : P, Mo et V) plus élevés comparés à ceux des matériaux PMo11V/HMS et PMo11V/CMI-1. Ainsi, on note 0,010 contre 0,005–0,009 pour P/Si, 0,066 contre 0,041 pour Mo/Si et 0,007 contre 0,003–0,006 pour V/Si. Ces différences sont dues au dépôt plus important des espèces hétéropolyanioniques à la surface plus petite du support SBA-15 et à ses parois plus épaisses comparées à celles des deux autres supports.
Les résultats du Tableau 5 montrent également que les rapports Mo/Si évalués à partir de l’XPS sont inférieurs à ceux obtenus par ICP (Tableau 1) (0,041 contre 0,132 pour PMo11V/HMS, 0,041 contre 0,139 pour PMo11V/CMI-1 et 0,066 contre 0,167 pour PMo11V/SBA-15). Ces résultats suggèrent une agglomération des espèces hétéropolyanioniques à la surface des supports, sous forme de petits agrégats non identifiés par DRX. Ces résultats peuvent être également expliqués par l’insertion de certaines molécules de l’hétéropolyacide dans les mésopores des supports comme observée par l’analyse BET [6].
3.8 Réactivité
L’oxydation du propène par l’oxygène moléculaire a été réalisée sur PMo11V à l’état massique, les supports HMS, CMI-1 et SBA-15 et sur 30 % PMo11V/support à 350 °C. Pour tous les essais catalytiques, il y a formation d’un produit non identifié qui se condense au niveau des colonnes. Cela conduit à un bilan de carbone inférieur à 100 %. Il a été rapporté dans la littérature que le bilan de carbone n’est jamais atteint pour les faibles conversions à cause de la faible exactitude des résultats analytiques [59].
Les résultats obtenus après cinq heures de réaction sont portés dans le Tableau 6. PMo11V à l’état massique et les supports, HMS, CMI-1 et SBA-15, conduisent à des conversions inférieures à 4 % et les COx sont les seuls produits observés. Supporté, PMo11V conduit à des conversions beaucoup plus élevées avec 22,8 % pour HMS, 24,5 % pour CMI-1 et 24,6 % pour SBA-15. La distribution des produits est peu sensible à la nature du support avec une légère amélioration des performances catalytiques de l’acide en présence de la HMS avec ∼ 39 % contre ∼ 45 et 46 % de sélectivité en COx, ∼ 16 % contre 12 et 11 % de sélectivité en produits oxygénés (acétaldéhyde, acroléine et acide acétique) en présence de CMI-1 et SBA-15, respectivement.
Oxydation du propène par l’oxygène moléculaire sur HMS, CMI-1, SBA-15 et 30 % PMo11V/support à 350 °C après cinq heures de réactiona.
| Catalyseur | Conversion (%) | Sélectivités (%) | |||
| COxd | Acétaldéhyde | Acroléine | Acide acétique | ||
| HMSb | 2,6 | 1,4 | 0,0 | 0,0 | 0,0 |
| CMI-1b | 4,1 | 1,5 | 0,0 | 0,0 | 0,0 |
| SBA-15b | 3,2 | 1,8 | 0,0 | 0,0 | 0,0 |
| PMo11Vc | 3,2 | 5,4 | 0,0 | 0,0 | 0,0 |
| PMo11V/HMSc | 22,8 | 39,4 | 7,2 | 4,0 | 4,3 |
| PMo11V/CMI-1c | 24,5 | 44,5 | 6,5 | 3,4 | 2,5 |
| PMo11V/SBA-15c | 24,6 | 46,0 | 5,6 | 3,2 | 2,5 |
a Mélange réactionnel : C3H6 : 3 mL min−1, O2 : 6 mL min−1, He : 21 mL min−1.
b Catalyseur ; 210 mg.
c Catalyseur ; 300 mg.
d COx ; CO + CO2.
Ces résultats confirment l’importance de la présence d’un support mésoporeux de grande surface spécifique dans l’amélioration des propriétés catalytiques de l’hétéropolyacide, H4PMo11VO40. Par ailleurs, la différence dans les propriétés texturales et morphologiques des trois supports ne semble pas avoir une grande influence sur les performances catalytiques de l’acide vu que les conversions et la sélectivité en produits oxygénés sont du même ordre de grandeur (23−25 % et 11−16 %, respectivement). La légère différence de comportement catalytique des trois systèmes semble être liée au nombre de sites actifs, espèces hétéropolyanioniques de surface (SiOH2+)(H3PMo11VO40−), plus important à la surface de la HMS, qui est plus élevée comparée à celles des 2 autres supports (HMS [650 m2/g] > CMI-1[610 m2/g] > SBA-15 [437 m2/g]).
3.9 Caractérisation des matériaux après tests catalytiques
La Fig. 7 présente les diffractogrammes de l’acide, H4PMo11VO40, à l’état massique avant et après cinq heures sous flux réactionnel à 350 °C. Après réaction, le diffractogramme obtenu montre une décomposition de l’acide en une phase différente de celle correspondant à l’oxyde de molybdène MoO3 [JCPDS 89–5108], suggérant une décomposition partielle du PMo11V.
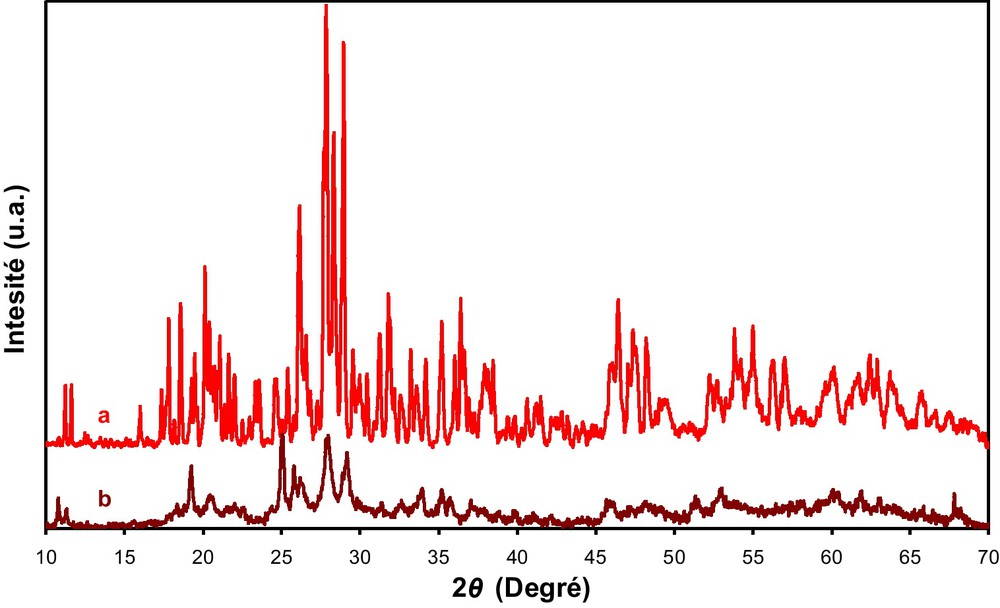
Diffractogrammes RX de PMo11V avant (a) et après (b) test catalytique pendant cinq heures à 350 °C.
L’analyse par DRX de PMo11V/support (30 %) montre des diffractogrammes similaires avec un large pic entre 2θ = 15 et 40° assigné à la silice amorphe (Fig. 8), ce qui ne permet pas de tirer une conclusion quant à la décomposition de l’acide pendant le test catalytique.
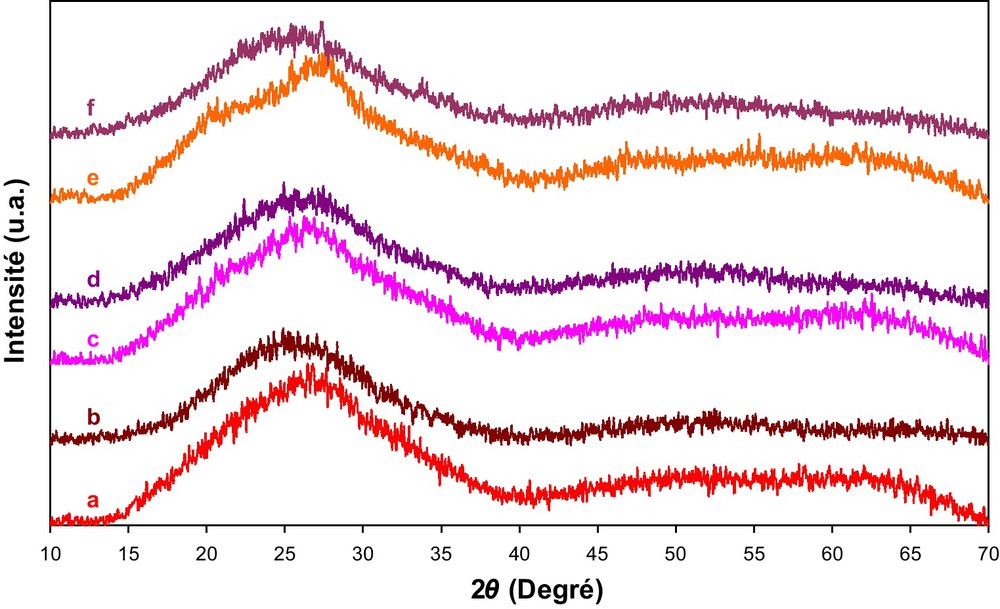
Diffractogrammes RX en poudre des catalyseurs PMo11V/HMS avant (a), après (b), PMo11V/CMI-1 avant (c), après (d) et PMo11V/SBA-15 avant (e) et après (f) tests catalytiques pendant cinq heures à 350 °C.
Les Fig. 9 et 10 montrent les spectres DRIFT de l’acide H4PMo11VO40 à l’état massique et supporté sur HMS avant et après cinq heures de test catalytique à 350 °C.
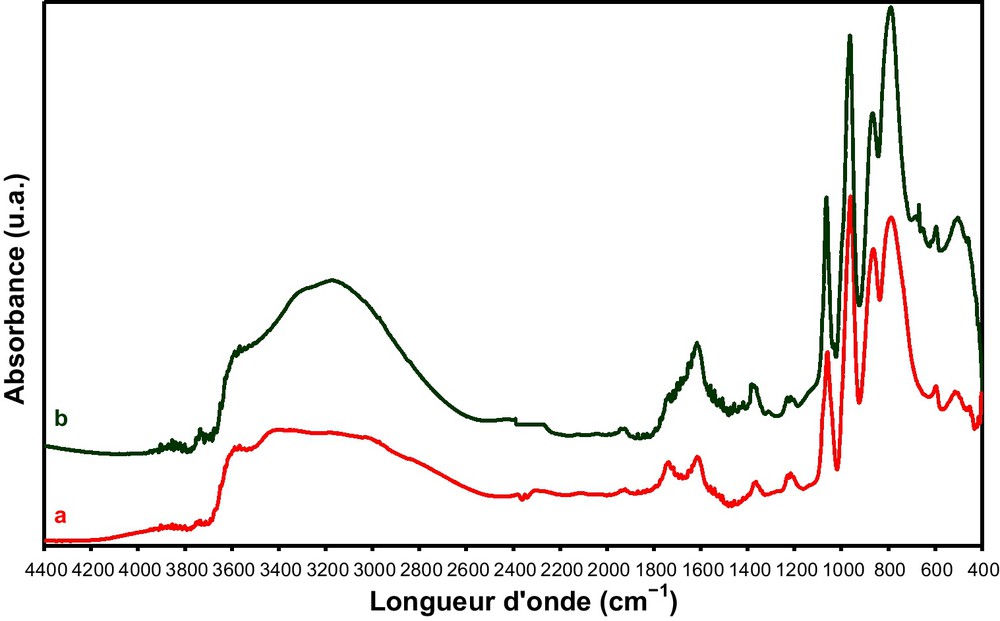
Spectres DRIFT du PMo11V avant (a) et après (b) test catalytique pendant cinq heures à 350 °C.
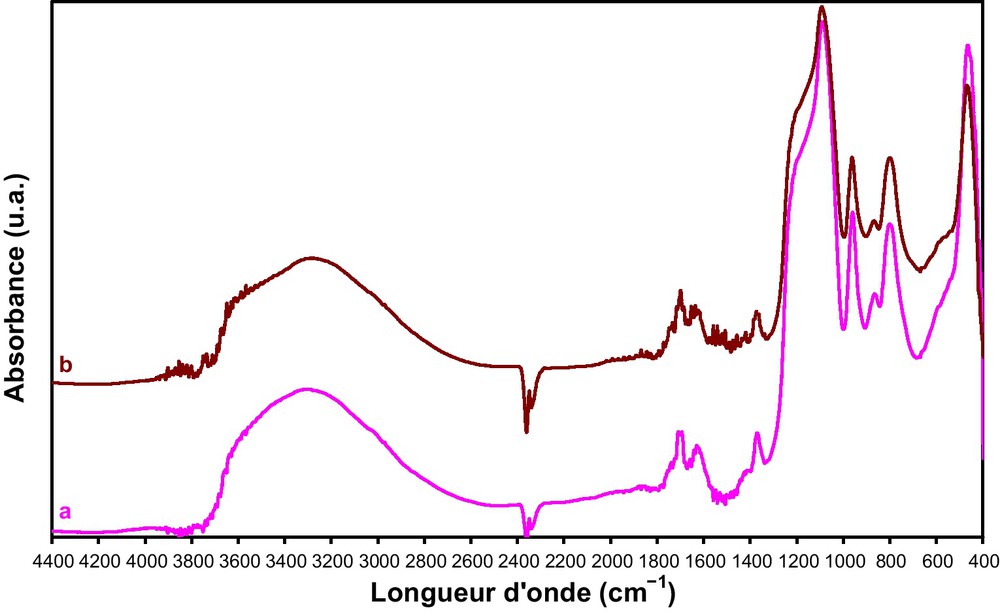
Spectres DRIFT du PMo11V/HMS avant (a) et après (b) test catalytique pendant cinq heures à 350 °C.
Les spectres DRIFT du PMo11V avant et après réaction sont similaires, indiquant que la structure de Keggin est maintenue après test catalytique. Cependant, une bande de faible intensité apparaît à ∼ 670 cm−1 assignée aux espèces V2O5 réduites [60]. Ces observations suggèrent que le vanadium migre de l’anion de Keggin vers l’extérieur sans détruire sa structure. Il a été rapporté dans plusieurs travaux, que le vanadium de l’anion de Keggin, [PMo11VO40]4−, sort de la structure du polyanion (position anionique) et se met en position de contre-ion dans le polyoxométallate lors d’un traitement thermique [61–63].
Les spectres DRIFT de PMo11V/HMS, avant et après réaction sont également identiques, montrant que la structure de Keggin n’a pas été détruite durant la réaction. Des résultats analogues ont été observés avec les supports CMI-1 et SBA-15.
Les résultats de la caractérisation des échantillons par spectroscopie DRIFT montrent que l’hétéropolyacide H4PMo11VO40, est stable après test catalytique, contrairement à l’analyse par DRX où une décomposition partielle est observée. Cette différence est probablement liée à l’aspect plus sensible de la première méthode ou à la recomposition de l’anion de Keggin après broyage avec le KBr. Il a été signalé que l’anion de Keggin pouvait se reconstituer en présence de traces d’eau [64].
Contrairement au PMo11V à l’état massique, les analyses par spectroscopie DRIFT et aussi la diffraction des RX de l’acide supporté, après oxydation du propène à 350 °C, ont montré la conservation de la structure de Keggin, indiquant que le support stabilise l’anion de Keggin, en accord avec les résultats de l’analyse thermique.
En spectroscopie Raman, la couleur foncée des échantillons, traduisant leur état réduit après test catalytique ne permet pas leur analyse. Le rayon incident est absorbé par l’échantillon réduit [65,66].
Les énergies de liaison de Mo 3d et V 2p3/2 avant et après cinq heures de test catalytique à 350 °C sont portées dans le Tableau 7. Une variation dans la valeur de E.L du V 2p3/2 de 0,5 à 0,8 eV est observée après réaction pour tous les systèmes catalytiques, suggérant la présence de vanadium avec des états d’oxydation inférieurs à V. Ces résultats montrent que les sites actifs dans l’oxydation du propène sont les espèces vanadium et non les espèces molybdène, résultat en accord avec ce qui a été rapporté par plusieurs auteurs [58,67,68].
Énergies de liaison (eV) avant et après cinq heures de test catalytique à 350 °C.
| PMo11V | PMo11V/HMS | |
| Mo 3da | 233,3 | 233,3 |
| Mo 3db | 233,2 | 233,3 |
| V 2pa | 518,1 | 517,2 |
| V 2pb | 517,3 | 516,7 |
a Avant réaction.
b Après réaction.
Dans le Tableau 8 sont portées les fractions molaires (%) de surface des spectres individuels des catalyseurs avant et après réaction.
Fractions molaires (%) de surface des spectres individuels des catalyseurs avant et après réaction.
| HMS | PMo11V/HMS | |
| O 1sa | 62,7 | 63,5 |
| O 1sb | 67,9 | 62,1 |
| C 1sa | 1,7 | 2,8 |
| C 1sb | 3,3 | 5,3 |
| Si 2pa | 35,6 | 32,2 |
| Si 2pb | 28,8 | 30,3 |
| P 2pa | − | 0,1 |
| P 2pb | − | 0,2 |
| Mo 3da | − | 1,3 |
| Mo 3db | − | 2,0 |
| V 2pa | − | 0,1 |
| V 2pb | − | 0,1 |
a Avant réaction.
b Après réaction.
Les quantités d’oxygène de surface varient légèrement après oxydation du propène en présence de PMo11V supporté, contrairement au support, HMS (62,1 contre 63,5 % et 67,9 contre 62,7 %, respectivement). Tandis que les quantités de carbone de surface augmentent après réaction (5,3 contre 2,8 % pour PMo11V/HMS et 3,3 contre 1,7 % pour la HMS). Cet excès de carbone confirmerait la formation du produit non identifié et sa condensation à la surface des catalyseurs. Ce résultat permettrait par ailleurs d’expliquer le bilan de carbone non atteint (< 100 %).
4 Conclusion
Les techniques d’analyse ont montré que la structure mésoporeuse et la morphologie des matériaux HMS, CMI-1 et SBA-15 sont différentes. La présence de l’hétéropolyacide H4PMo11VO40 modifie les propriétés texturales (diminution de surface spécifique et de volume poreux) de ces matériaux sans détruire leur structure (taille des pores inchangée). Les analyses spectroscopiques (FT–IR, DRIFT) ont montré que la structure de Keggin de l’acide H4PMo11VO40 est préservée après son imprégnation sur les différents supports.
L’étude sur la réactivité de l’hétéropolyacide, H4PMo11VO40 dans l’oxydation du propène, a montré l’importance de l’utilisation d’un support mésoporeux de grande surface spécifique dans l’amélioration de ses propriétés catalytiques (meilleures activité et sélectivités en produits oxygénés valorisables, acétaldéhyde, acroléine et acide acétique).
L’interaction support–hétéropolyacide conduit à la formation d’espèces hétéropolyanioniques de surface (SiOH2+)(H3PMo11VO40−) bien dispersées, plus stables que les espèces acides, H4PMo11VO40, et qui semblent être les sites actifs.
Les analyses par spectroscopie DRIFT et diffraction des RX de l’hétéropolyacide supporté, après oxydation du propène à 350 °C, confirment la stabilité accrue des HPAs supportés.