

1 Introduction
Imidazole and its derivatives as a major kind of heterocyclic compounds play an important role in biochemistry [1–5], material chemistry including luminescent material [6,7] and proton conductivity material [8,9], medicinal chemistry [10–14] and the determination of heavy metal ion [15] and albumin in human serum samples [16] due to their structure nature. Therefore, effective methods for preparation of imidazole derivatives are of great interest in organic synthesis. The preparation for imidazole derivatives were usually based on the condensation of alkyl (aryl)-diones or 2-hydroxy-1,2-diphenylethanone with aldehydes, or β-carbonyl-N-acyl-N-alkylamines in the present of ammonium acetate, ammonia or primary amines in different solvent by various chemical methods like refluxing [17–20], solid-phase synthesis [21], heating in ionic liquid [22], sodium dihydrogen phosphate acted as catalyst [23], MW/silica-gel [24], MW/Al2O3 [25], etc. However, many of the methods reported above suffer from one or more disadvantages such as complex operation process, high solvent consumption and longer reaction time. Moreover, alkyl- or aryl-diones and aldehydes, benzoin and aldehydes were usually used as starting materials in these methods. Few methods were applied to the synthesis of furly-substituted imidazoles. Because the furan ring possess the inherent chemical properties and disadvantages, such as rich electron, lower conjugation energy, easily ring-opening in Brönsted acid, easily becoming yellow in oxygen and so on. Therefore, it is very significant to study and develop new scaleable synthetic routes able to construct fused imidazole derivatives in high yields. Especially imidazoles with furan rings which may extend the application of imidazole derivatives. For this purpose, microwave heating was introduced into the synthesis by allowing us to obtain rapid, reproducible, and scalable processes to synthesize new molecules in high yield. This methodology can facilitate the cyclization, and is well-suited for synthesizing the compounds with heterocyclic rings. In fact, traditional heating reactions often require high temperature for many hours or even days in the present of organic solvent, whereas similar or higher yields and cleaner process can be obtained by microwave heating for a few minutes [13,24–28]. Our work focused on the synthesis of 2-substituted-4,5-di(2-furyl)imidazoles from 1,2-di(furan-2-yl)-2-oxoethyl carboxylates. It is known that the use of alumina or silica-gel in a solvent-free fashion greatly eases work-up procedures. To the best of our knowledge, the microwave-assisted and starting from 1,2-di(furan-2-yl)-2-oxoethyl carboxylates version of this reaction has not been reported, although several papers describing other microwave-assisted syntheses of imidazoles with benzene rings [24,25] and other synthesis of few furly-substituted imidazoles by heating in ionic liquid [22] have been published.
Herein, we wish to report a facile and environmentally-friendly method for the synthesis of trisubstituted imidazoles containing furan rings from 1,2-di(furan-2-yl)-2-oxoethyl carboxylates and using alumina as a safe, inexpensive solid support under solvent-free and microwave-assisted conditions (Scheme 1). In addition, when the method was used to synthesize trisubstituted imidazoles containing benzene rings, the desired products were also obtained. However, the yields were appreciably lower than that of trisubstituted imidazoles containing furan rings.
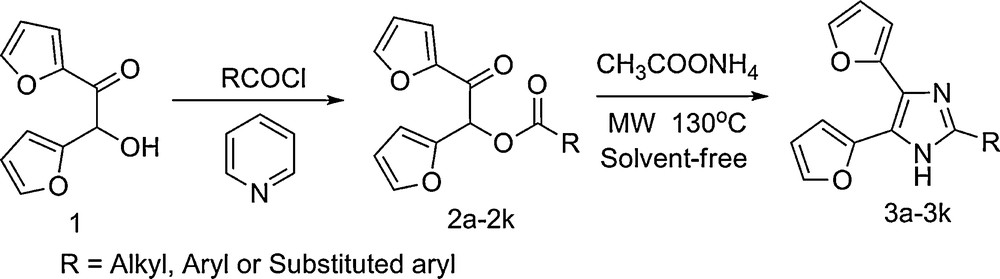
The pathway of trisubstituted imidazoles containing furan rings.
2 Experimental
Furoin (1,2-di(furan-2-yl)-2-hydroxyethanone) was generated from furfural according to the reported procedure [29]. All reagents were of analytical-reagent grade and used without further purification. All reported yields were isolated yields. All melting points were determined on a XT − 4 melting point apparatus and were uncorrected. 1H and 13C NMR spectra were measured using a Varian Mercury-300 NMR spectrometer or a Bruker AVANCE-500 NMR spectrometer and with TMS as an internal standard. MS and HRMS were collected with an Agilent HP1100/6890 LC/MS spectrometer and an Agilent1290-micrOTOF Q II spectrometer, respectively. Elemental analyses were determined with a FlashEA1112 elemental analyzer. FT-IR spectra were obtained as KBr pellets using an IRAffinity-1 instrument in the range of 500–3500 cm−1. A MCL-3-type microwave reactor with a thermometer was used in all experiments (Microwave chemical lab, College of Electronics and information Engineering, Sichuan University Sichuan, China). The CL emission was detected by an ultra-weak luminescence analyzer (type BPCL manufactured at the Institute of Biophysics, Chinese Academy of Sciences, Beijing, China). The acquisition and treatment of data were performed with BPCL software running under Windows XP.
2.1 Experimental procedures for compounds (2a–2k)
1,2-di(furan-2-yl)-2-oxoethyl carboxylates were synthesized according to the literature [30]. Furoin (5 mmol, 0.96 g) and acyl chlorides (5.5 mmol) were mixed in the three-neck flask. The mixture was heated to 30−40 °C in water bath with stirring for 15 min. Pyridine (3 mL) was slowly dropped into the mixture and stirred at the above temperature until the reaction was over by TLC analysis. Then the mixture was cooled, filtered and the residue was washed with iced ethanol (3 × 5 mL), water (3 × 10 mL), and recrystallized from 95% ethanol to give the pure product.
2.2 Experimental procedures for the synthesized compounds (3a–3k) under solvent−free condition
Typical procedure for the synthesis of trisubstituted imidazoles as follows: a mixture of alumina (0.8 g, 7.84 mmol) or silica-gel (0.5 g, 8.32 mol) ammonium acetate (0.23 g, 3 mmol) and 1,2-di(furan-2-yl)-2-oxoethyl carboxylates (1 mmol) was fully ground in a mortar, and then transferred to a 50 mL dried two-neck round-bottomed flask containing a thermometer and condenser, and heated with microwave irradiation for 5 min (the reaction temperature was 130 °C when the reaction was over.) or conventional heating at 130 °C for 40–90 min. The procedure was repeated twice. The reaction progress was monitored by TLC on Silufol-254 plates. When the microwave-assisted reaction was over, the residue was cooled to room temperature and was directly purified by column chromatography on Chemapol (200–300 mesh) silica gel (eluent, petroleum ether/ethyl acetate = 3/1) to give the desired products.
2.3 Physical and spectroscopic data
4,5-di(furan-2-yl)-2-phenyl-1H-imidazole (3a) Mp: 197–198 °C (lit. [21] 218 °C). Yield: 74.5%. IR (KBr): ν (cm−1) 3118, 3054, 1599, 1551, 1481, 1404, 1233, 1090, 886, 732, 590. 1H NMR (300 MHz, CDCl3): δ (ppm): 7.92 (dd, J = 8.2, 1.5 Hz, 2H), 7.57–7.33 (m, 5H), 6.99 (d, J = 3.3 Hz, 2H), 6.53 (dd, J = 3.4, 1.8 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 146.74, 129.64, 129.52, 129.23, 125.96, 112.13, 108.00. MS, m/z: (M + H)+ 277.9, (M)+ 276.9 (Calcd 276.29). Anal. calcd for C17H12N2O2: C, 73.90; H, 4.38; N, 10.14. Found: C, 73.62; H, 4.34; N, 9.95.
4,5-di(furan-2-yl)-2-(4-methylphenyl)-1H-imidazole (3b) Mp: 217–219 °C. Yield: 75.5%. IR (KBr): ν (cm−1) 3125, 3017, 2917, 2853, 1599, 1530, 1493, 1433, 1374, 1262, 1200, 1021, 912, 823, 732, 590. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.79 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 1.2 Hz, 2H), 7.19(s, 2H), 6.97 (d, J = 3.2 Hz, 2H), 6.64–6.35 (m, 2H), 2.34(s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 146.74, 139.70, 129.98, 126.84, 125.80, 112.16, 107.89, 21.77. MS, m/z: (M + H)+ 291.2 (Calcd 291.31). Anal. calcd for C18H14N2O2: C, 74.47; H, 4.86; N, 9.65. Found: C, 74.52; H, 4.76; N, 9.95.
4,5-di(furan-2-yl)-2-(4-chlorophenyl)-1H-imidazole (3c) Mp: 216–218 °C. Yield: 81.6%. IR (KBr): ν (cm−1) 3115, 3052, 1598, 1525, 1483, 1432, 1374, 1260, 1201, 1011, 835, 736, 590. 1H NMR (300 MHz, CDCl3): δ (ppm): 7.87 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 1.7 Hz, 2H), 7.42 (d, J = 8.6 Hz, 2H), 7.01 (d, J = 3.2 Hz, 2H), 6.54 (dd, J = 3.4, 1.8 Hz, 2H). 13C NMR (125 MHz, DMSO): δ (ppm) 145.44, 141.99, 135.56, 129.53, 127.97, 127.15, 112.22, 108.25. MS, m/z: (M + 2 + H)+ 313.8, (M + 2)+ 312.8, (M + H)+ 311.8, (M)+ 310.8 (Calcd 310.7). Anal. calcd for C17H11ClN2O2: C, 65.71; H, 3.57; N, 9.02. Found: C, 65.46; H, 3.65; N, 8.83.
4,5-di(furan-2-yl)-2-(2-furylvinyl)-1H-imidazole (3d) Mp: 213–215 °C. Yield: 89.7%. IR (KBr): ν (cm−1) 3121, 3027, 1625, 1482, 1425, 1370, 1255, 1201, 1021, 885, 733, 589. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.46 (d, J = 1.7 Hz, 2H), 7.37–7.29 (m, 2H), 7.00 (d, J = 3.4 Hz, 2H), 6.83 (d, J = 16.2 Hz, 1H), 6.49 (dd, J = 3.4, 1.8 Hz, 2H), 6.40–6.34 (m, 2H).13C NMR (125 MHz, CDCl3): δ (ppm) 152.38, 147.03, 145.58, 143.54, 142.00, 120.98, 113.11, 112.34, 112.19, 111.49, 108.31. MS, m/z: (M)+ 292.9, (M + H)+ 293.9 (Calcd 293.30). Anal. calcd for C17H12N2O3: C, 69.86; H, 4.14; N, 9.58. Found: C, 69.68; H, 4.29; N, 9.30.
4,5-di(furan-2-yl)-2-(1-chloro-2,2-dimethylethyl)-1H-imidazole (3e) Mp: 186–187 °C. Yield: 48.9%. IR (KBr): ν (cm−1) 3444, 3130, 3026, 2983, 2940, 2815, 1602, 1544, 1517, 1473, 1410, 1367, 1215, 1190, 1074, 1011, 976, 885, 812, 738, 593. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.48 (s, 2H), 6.88 (s, 2H), 6.50 (dd, J = 3.4, 1.8 Hz, 2H), 3.81 (s, 2H), 1.56 (s, 6H). 13C NMR (125 MHz, DMSO): δ (ppm) 152.23, 141.73, 112.29, 111.77, 107.60, 54.85, 38.51, 25.45. MS, m/z: (M + H)+ 291.8 (M)+ 290.8 (Calcd 290.74); Anal. calcd for C15H15ClN2O2: C, 61.97; H, 5.20; N, 9.64. Found: C, 61.77; H, 5.18; N, 9.66.
4,5-di(furan-2-yl)-2-methyl-1H-imidazole (3f) Mp: 164–166 °C. Yield: 60.4%. IR (KBr): ν (cm−1) 3415, 3131, 3000, 2902, 2786, 1610, 1560, 1430, 1272, 1216, 1159, 1091, 1022, 927, 886, 912, 813, 724, 594. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.45 (d, J = 1.1 Hz, 2H), 6.90 (d, J = 3.3 Hz, 2H), 6.50 (dd, J = 3.3, 1.8 Hz, 2H), 2.47 (s, 3H). 13C NMR (125 MHz, DMSO): δ (ppm) 147.73, 145.36, 141.55, 123.55, 111.99, 107.20, 14.19. MS, m/z: (M)+ 214.8 (Calcd 214.22). Anal. calcd for C12H10N2O2: C, 67.28; H, 4.71; N, 13.08. Found: C, 67.43; H, 4.65; N,13.10.
4,5-di(furan-2-yl)-2-(4-nitrophenyl)-1H-imidazole (3g) Mp: 208–210 °C (lit. [21] 208 °C). Yield: 86.2%. IR (KBr): ν (cm−1) 3428, 3095, 3032, 1600, 1510, 1340, 1250, 1201, 1110, 1080, 991, 906, 886, 850, 798, 731, 629. 1H NMR (300 MHz, CDCl3): δ (ppm) 8.29 (d, J = 8.9 Hz, 2H), 8.09 (d, J = 8.9 Hz, 2H), 7.53 (d, J = 1.3 Hz, 2H), 7.04 (s, 2H), 6.56 (dd, J = 3.5, 1.8 Hz, 2H). 13C NMR (125 MHz, DMSO): δ (ppm) 147.71, 144.93, 136.31, 127.12, 125.05, 112.63. MS, m/z: (M + H)+ 322.9, (M)+ 321.8 (Calcd 321.29). Anal. calcd for C17H11N3O4: C, 63.55; H, 3.45; N, 13.08. Found: C, 63.25; H, 3.52; N, 12.98.
3-(4,5-di(furan-2-yl)-1H-imidazol-2-yl)pyridine (3h) Mp: 244–246 °C. Yield: 82.5%. IR (KBr): ν (cm−1) 3440, 3150, 3106, 3045, 1600, 1545, 1523, 1474, 1450, 1373, 1264, 1210, 1091, 1010, 962, 909, 884, 731, 594. 1H NMR (300 MHz, DMSO): δ (ppm) 9.27 (s, 1H), 8.61 (d, J = 3.5 Hz, 1H), 8.47–8.42 (m, 1H), 7.80 (d, J = 1.1 Hz, 2H), 7.55 (dd, J = 8.1, 4.9 Hz, 1H), 6.89 (d, J = 3.0 Hz, 2H), 6.64 (dd, J = 3.3, 1.8 Hz, 2H). 13C NMR (125 MHz, DMSO): δ (ppm): 149.82, 147.17, 144.41, 143.32, 134.14, 126.65, 124.83, 112.57, 108.86. MS, m/z: (M + H)+ 278.8 (Calcd 278.29). Anal. calcd for C16H11N3O2: C, 69.31; H, 4.00; N, 15.15. Found: C, 69.05; H, 4.12; N, 15.30.
4,5-di (furan-2-yl)-2-undecyl-1H-imidazole (3i) Mp: 80–82 °C. Yield: 56.0%. IR (KBr): ν (cm−1) 3423, 3120, 3024, 2920, 2850, 1600, 1550, 1512, 1470, 1415, 1380, 1270, 1210, 1194, 1160, 1080, 1020, 912, 883, 806, 733, 660, 593. 1H NMR (300 MHz, DMSO): δ (ppm) 12.30 (s, 1H), 7.72 (s, 2H), 6.74 (s, 2H), 6.58 (s, 2H), 2.63 (s, 2H), 1.67 (s, 2H), 1.24 (s, 16H), 0.85 (s, 3H). 13C NMR (125 MHz, DMSO): δ (ppm) 150.11, 142.52, 132.58, 129.53, 112.39, 107.33, 32.16, 30.88, 29.87, 29.79, 29.57, 29.47, 28.81, 28.46, 19.52, 14.81. MS, m/z: (M + H)+ 355.1. (Calcd 354.49). Anal. calcd for C22H30N2O2: C, 74.54; H, 8.53; N, 7.90. Found: C, 74.26; H, 8.48; N, 7.70.
2,4,5-tri (furan-2-yl)-1H-imidazole (3j) Mp: 196–197 °C. Yield: 73.3%. IR (KBr): ν (cm−1) 3110, 3420, 1400, 1200, 1420, 1540, 885, 733, 590. 1H NMR (500 MHz, CDCl3): δ (ppm) 10.48 (s, 1H),7.42 (s, 2H), 7.36 (s, 1H, furan-5H), 6.92 (d, J = 3.3 Hz, 3H), 6.46 (dd, J = 3.0, 1.7 Hz, 2H), 6.41 (dd, J = 3.1, 1.6 Hz, 1H). 13C NMR (125.5 MHz, CDCl3) δ (ppm) 147.2, 145.2, 143.1, 141.9, 139.5, 112.4, 112.1, 108.9, 108.1. MS, m/z: (M + H)+ 267.3, (M)+ 266.9, (Calcd 266.25). Anal. calcd for C15H10N2O3: C, 67.67; H, 3.79; N, 10.52. Found: C, 67.57; H, 3.73; N, 10.46.
4,5-di(furan-2-yl)-2-styryl-1H-imidazole (3k) Mp: 209–211 °C. Yield: 90.3%. IR (KBr): ν (cm−1) 3420, 3110, 2970, 2920, 1626, 1540, 1501, 1430, 1372, 1260, 1200, 1160, 1070, 1020, 987, 962, 912, 883, 806, 733, 685, 590. 1H NMR (500 MHz, DMSO): δ (ppm) 12.75 (s, 1H), 7.78 (d, J = 48.9 Hz, 2H), 7.59 (d, J = 7.5 Hz, 2H), 7.52 (d, J = 16.5 Hz, 1H), 7.41 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.06 (d, J = 16.5 Hz, 1H), 6.97 (s, 1H), 6.66 (dd, J = 44.5, 31.3 Hz, 3H). 13C NMR (125 MHz, DMSO): δ (ppm): 146.66, 137.09, 129.78, 127.45, 117.37, 112.86, 112.32, 108.86, 107.61. MS, m/z: (M + H)+ 303.0 (Calcd 303.34). Anal. calcd for C19H14N2O2: C, 75.58; H, 4.67; N, 9.27. Found: C, 75.82; H, 4.81; N, 9.45.
4,5-di(phenyl)-2-(2-furylvinyl)-1H-imidazole (3l) Mp: 194–196 °C. Yield: 83.3%. IR (KBr) ν: 3060, 2960, 1600, 1580, 1480, 1450, 1320, 1260, 1200, 1070, 1010, 968, 883, 764, 696, 592 cm−1. 1H NMR (CDCl3, 500 MHz) δ: 7.51 (d, J = 7.2 Hz, 4H), 7.39 (d, J = 1.1 Hz, 1H), 7.32 (t, J = 7.3 Hz, 4H), 7.28 (d, J = 7.2 Hz, 2H), 7.22 (d, J = 16.1 Hz, 1H), 6.85 (d, J = 16.1 Hz, 1H), 6.41 (dd, J = 3.2, 1.8 Hz, 1H), 6.38 (d, J = 3.3 Hz, 1H); 13 C NMR (DMSO, 126 MHz) δ: 152.29, 145.16, 142.80, 132.50, 129.15, 128.56, 127.84, 127.47, 119.36, 113.69, 111.87, 110.39; HRMS, m/z: (M + H)+ 313.1334 (Calcd 313.1335).
4,5-di-phenyl-2-styryl-1H-imidazole (3m) Mp: 227–229 °C. Yield: 77.6%. IR (KBr) ν: 3030, 2960, 1600, 1500, 1450, 1200, 1070, 1030, 958, 764, 696, 602 cm−1. 1H NMR (DMSO, 500 MHz) δ: 12.60 (s, 1H), 7.60 (d, J = 7.4 Hz, 2H), 7.48 (d, J = 22.8 Hz, 5H), 7.44–7.18 (m, 9H), 7.10 (d, J = 16.5 Hz, 1H); 13 C NMR (DMSO, 126 MHz) δ: 145.71, 136.91, 130.45, 129.37, 128.92, 128.48, 128.03, 126.93, 117.68; HRMS, m/z: (M + H)+ 323.1543 (Calcd 323.1543).
3 Results and discussion
3.1 Synthesis of compounds
In our previous work, many subsequent products correlated with furfural had been synthesized in our laboratory [29–32]. In the present work, eleven 1,2-di(furan-2-yl)-2-oxoethyl carboxylates were readily synthesized by the esterification of furoin with 1.1 equivalent of acyl chlorides and using pyridine as catalyst in excellent yields (85.4–94.7%) under mild reaction conditions referring to our reported method [30] (Table 1).
Synthesis of 1,2-di(furan-2-yl)-2-oxoethyl carboxylates from furoin with acyl chlorides catalysed by pyridine.a
| Entry | R-COCl | Product | Yieldb (%) | Mp.(°C) |
| 1 | 2a | 94.1 | 78-79 | |
| 2 | 2b | 91.5 | 115-117 | |
| 3 | 2c | 94.3 | 96-98 | |
| 4 | 2d | 92.3 | 108-110 | |
| 5 | 2e | 85.4 | 95-97 | |
| 6 | 2f | 90.9 | 72-73 | |
| 7 | 2g | 94.7 | 117-118 | |
| 8 | 2h | 90.5 | 92-94 | |
| 9 | 2i | 88.2 | 54-55 | |
| 10 | 2j | 90.4 | 129-130 | |
| 11 | 2k | 92.6 | 105-107 |
a Reaction condition: 5 mmol Furoin, 5.5 mmol acyl chlorides, 3 mL pyridine at 35 °C.
b Isolated yield.
Subsequently, the synthesis of trisubstituted imidazoles containing furan ring was explored starting from 1,2-di(furan-2-yl)-2-oxoethyl carboxylates. Herein, the nucleophilic addition and self-cycloaddition reaction of 1,2-di(furan-2-yl)-2-oxoethyl carboxylates with 3.0 equivalent of ammonium acetate was readily carried out. Under otherwise similar experimental conditions, the effect of different heating conditions and solid support on the yield of the products is shown in Table 2. We successfully used alumina or silica-gel as solid support to synthesize the desired product in almost equal yields, and the yield is higher than that of the product obtained under conventional heating (over 9.0%) and under microwave heating (6.8–7.6% except product 3j) in the absence of solid support, respectively. This is because alumina and silica-gel possess the predominant ability to disperse reactant and microwave, together with possible catalysis which sufficiently promoted the reaction. The effect is consonant with that of solid support in literature [26]. Comparison of different heating conditions and solid supports showed that microwave heating on solid support alumina or silica-gel with the identical diameter (200–300 mesh) was more suitable for the synthesis of 3a, 3d, 3j and 3k from 2a, 2d, 2j and 2k (Table 2). Because the loading of solid supports is relative to their specific surface area, the optimum amounts for alumina and silica-gel are around 0.8 g (7.84 mmol) and 0.5 g (8.32 mmol), respectively. Alternatively, all the other compounds were synthesized under microwave heating on solid support alumina.
Optimization of the reaction conditions for the mode of heating and solid support.
| Products | Conventional syntheses (%) | Microwave heated syntheses | ||
| No solid support (%) | Alumina (%) | Silica-gel (%) | ||
| 3a | 64.8 | 67.7 | 74.5 | 75.2 |
| 3d | 80.3 | 82.2 | 89.7 | 89.0 |
| 3j | 63.5 | 72.4 | 73.3 | 73.8 |
| 3k | 81.1 | 82.7 | 90.3 | 89.4 |
In addition, the effect of microwave irradiation times on the yields of product was investigated by employing 1,2-di(furan-2-yl)-2-oxoethyl-4-nitrobenzoate and ammonium acetate on solid support alumina under microwave radiation in the absence of any organic solvent. It was observed that the yield of 4,5-di(furan-2-yl)-2-(4-nitrophenyl)-1H-imidazole (3h) was substantially increased under microwave irradiation for 5 min compared with that at shorter irradiation time (Fig. 1). However, the yield was not obviously enhanced when reaction time was further increased to 10 min and 15 min. Consequently, trisubstituted imidazoles containing furan rings were synthesized in moderate to excellent yield from 1,2-di(furan-2-yl)-2-oxoethyl carboxylates and ammonium acetate by the same method and heated for 5 min in microwave. No reaction was observed without heating. Furthermore, the yield of product was reduced when the reaction mixture was deficiently mixed with solid support.
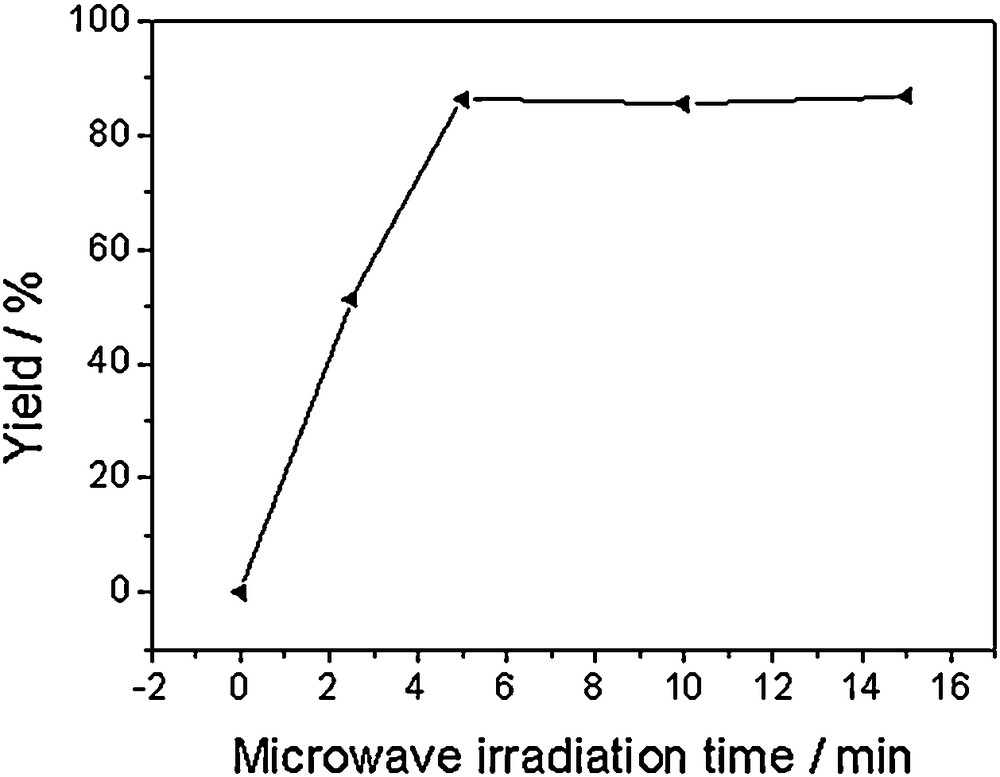
The effect of microwave irradiation time on yield.
In the previous literature [33], there were two products 1,3-oxazole and 1,3-imidazole which were obtained in approximately equal yield (about 40%) by traditional heating based on α-acyloxy ketones as raw materials and ammonium acetate in boiling glacial acetic acid. Besides, the substituted oxazoles were prepared in moderate yield by improving the above-mentioned synthesis procedures in the literature [34]. In our work, eleven trisubstituted imidazoles with furan rings were synthesized by using 1,2-di(furan-2-yl)-2-oxoethyl carboxylates under microwave radiation and solvent-free conditions (Table 3). Herein, only a few trisubstituted oxazoles reported in the literature [33–35] were found in the synthesizing process of trisubstituted imidazoles. The results of microwave-assisted nucleophilic addition from a range of 1,2-di(furan-2-yl)-2-oxoethyl carboxylates 2a–k with ammonia to obtain the products 3a–k are listed in Table 3. In all examples, excellent yields were obtained after 5 min. The nucleophilic addition and self-cycloaddition reaction of 1,2-di(furan-2-yl)-2-oxoethyl carboxylates with 3.0 equivalent of ammonium acetate was readily carried out. The structures of imidazole derivatives were confirmed by elemental analysis, IR, 1H and 13C NMR and MS analysis. The NHRCN protons of compounds exhibit resonances at 9.27–12.75 ppm (DMSO as solvent) or not exhibit resonances (CD3Cl as solvent), while the resonances for the corresponding NHRCN carbon atoms are observed at 136–145 ppm.
Synthesis of trisubstituted imidazoles containing furan ring from the reaction of 1,2-di(furan-2-yl)-2-oxoethyl carboxylates with ammonia dispersed by Al2O3 under solvent-free and microwave heating conditions.a
| Entry | R | Time (min) | Product | Yieldb (%) |
| 1 | 5 | 3a | 74.5 | |
| 2 | 5 | 3b | 75.5 | |
| 3 | 5 | 3c | 81.6 | |
| 4 | 5 | 3d | 89.7 | |
| 5 | 5 | 3e | 48.9 | |
| 6 | 5 | 3f | 60.4 | |
| 7 | 5 | 3g | 86.2 | |
| 8 | 10 | 3h | 82.5 | |
| 9 | 5 | 3i | 56.0 | |
| 10 | 5 | 3j | 73.3 | |
| 11 | 5 | 3k | 90.3 | |
| 12c | 5 | 3l | 83.3 | |
| 13c | 5 | 3m | 77.6 |
a Reaction condition: 1,2-di(furan-2-yl)-2-oxoethyl carboxylates (1 mmol), ammonium acetate (3 mmol).
b The yield purified by column chromatography.
c Reaction condition: 1,2-di-phenyl-2-oxoethyl carboxylates (1 mmol), ammonium acetate (3 mmol)
As shown in Table 3, it is clear that this approach could be applied to a broad range of 2-oxoethyl carboxylates. The results indicated that good yields were obtained when 2-oxoethyl carboxylates containing aromatic rings, heterocyclic rings and the longer conjugated substituted groups were used as starting materials (entries 1–4, 7, 8, 10 and 11), and increasing the conjugated chain, the yield of the products was enhanced (entries 4 and 11). However, when 2-oxoethyl aliphatic ester was used, the yield was lower (entries 5, 6 and 9). This is due to that the conjugated effect of the substituted aromatic rings and the longer conjugated substituted groups enhanced the stability of the hypothetic intermediates N-(2-amino-1,2-di-furan-2-vinyl)-amides(m), N-(1,2-di-furan-2-yl-2-oxo-ethyl)-amides(n) and the products. Besides, the electron-withdrawing effect of aromatic rings with electron-withdrawing group and pyridyl (entries 3, 7 and 8) made the electrophilicity of the carbon of carboxyl much stronger, and the self-cycloaddition easily perform. Therefore, the yield of products was high, whereas, when there is phenyl, 2-furyl and electron-donating group on the aromatic rings, better yields were obtained (entries 1–3 and 10), due to the fact that the electron-donating effect led to the self-cycloaddition becoming a little difficult. Simultaneously, the effect of substituted groups on the yield was also explained by the proposed possible mechanism (Scheme 2).
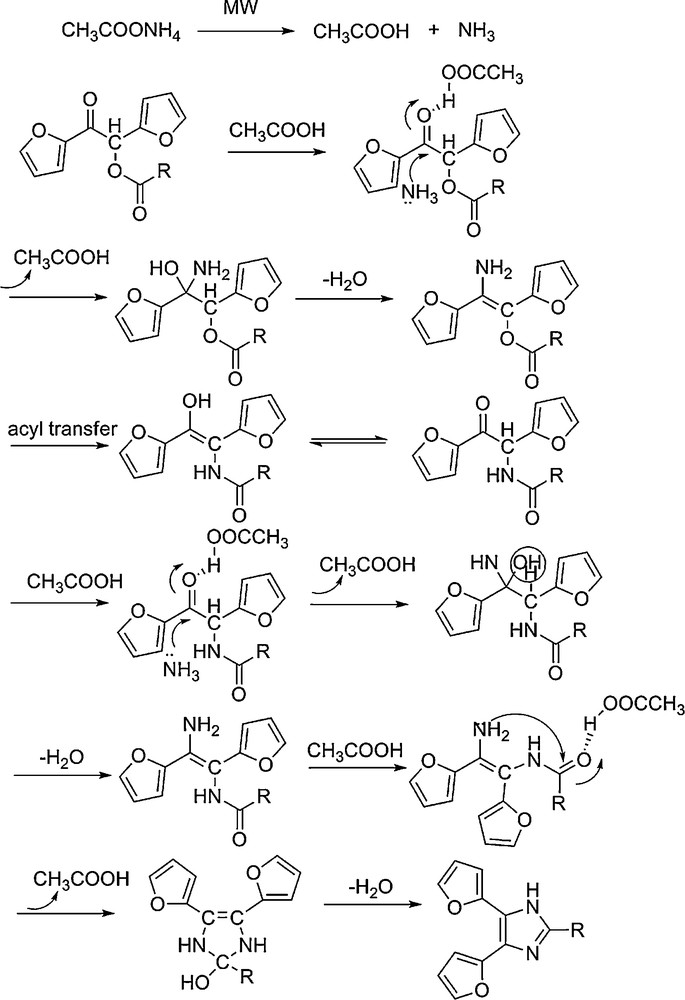
The possible mechanism to generate trisubstituted imidazoles.
The possible mechanism to generate trisubstituted imidazoles including furan rings from 1,2-di(furan-2-yl)-2-oxoethyl carboxylic esters is proposed, as shown in Scheme 2. The ammonium acetate decomposed to produce the catalyst acetic acid, which further forms hydrogen bond with the carboxyl group on 1,2-di (furan-2-yl)-2-oxoethyl carboxylates. As a result, the electrophilicity of the carbon of carboxyl was substantially enhanced, which accelerated the following nucleophilic addition and self-cycloaddition with ammonia to form the 1,2-di(furan-2-yl)-2-oxoethyl carboxylates–ammonia adduct and the final product. Based on the results described above, we can conclude that microwave-assisted one-pot procedure on the surface of alumina provides a simple and efficient method for the synthesis of trisubstituted imidazoles under solvent-free conditions. In addition, the method was applied to the synthesis of trisubstituted imidazoles with benzene rings, 4,5-dipenyl-2-styryl-1H-imidazole and 2-(furan-2-yl) vinyl-4,5-dipenyl-1H-imidazole were successfully synthesized in 83.3% and 77.6% (entries 12 and 13 in Table 3), respectively.
3.2 The determination of Cu2+ by CL reaction of the synthesized compounds
The compounds synthesized (3a and 3j) were applied to the determination of Cu2+ by the CL reaction with H2O2. The H2O2 could directly oxidize 3j/3a to produce CL emission in basic solution. The dynamic CL intensity-time profiles of the 3j/3a-H2O2 systems are shown in Fig. 2 (curves b and c). It has been found experimentally that the addition of Cu2+ into the system could induce significant enhancement of CL signal. The results are shown in Fig. 1 (curve d and e). So, Cu2+ was chosen to enhance the CL intensity of the 3j/3a-H2O2 system in the experiment. The effects of experimental conditions were investigated. The results are shown in Table 4. Under the optimal conditions, the relationship between enhanced CL intensity and the concentration of Cu2+ was obtained. The linear ranges of the calibration curves are 5.0 × 10−10–1.0 × 10−5 mol/L for 3j-H2O2-Cu2+ system, 1.0 × 10−10–5.0 × 10−6 mol/L for 3a-H2O2-Cu2+ system, and the corresponding detection limits are 2.7 × 10−10 and 6.5 × 10−11 mol/L, respectively. The proposed method has been used to determine trace amount of Cu2+.
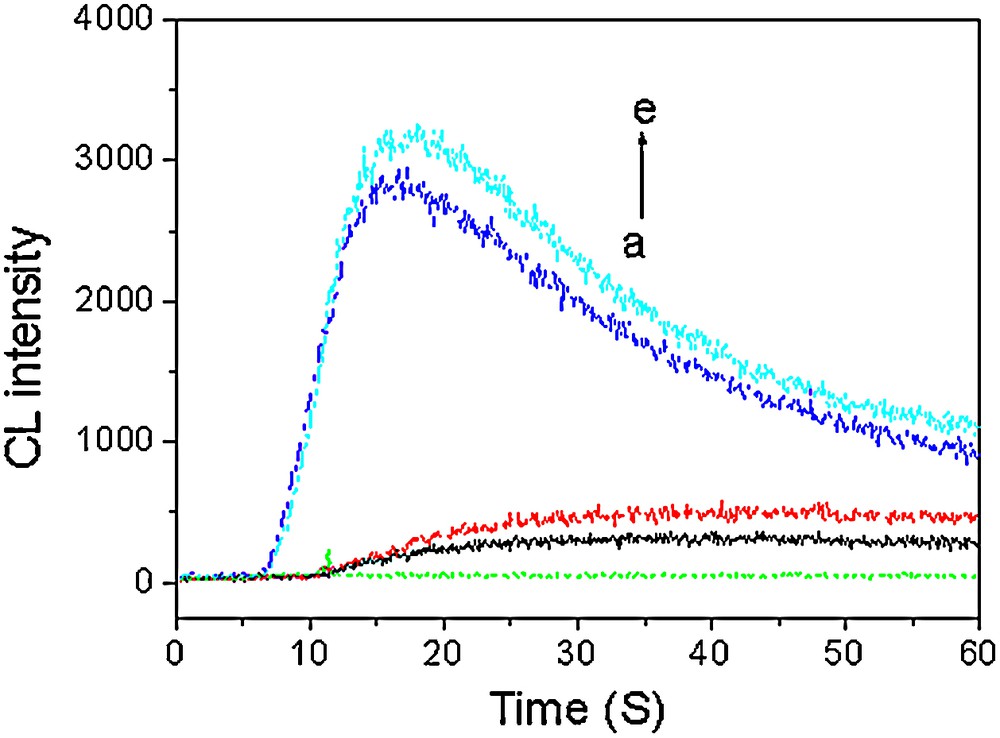
CL kinetic curves of different systems: a: H2O2: b: 3a-H2O2; c: 3j-H2O2; d: Cu2+-3a-H2O2; e: Cu2+-3j-H2O2 system. Concentration: 3a, 0.1 mmol/L; 3j, 0.1 mmol/L; NaOH, 0.1 mol/L; H2O2, 1 mol/L; Cu2+, 0.01 mmol/L.
Optimal conditions of 3j/3a-H2O2-Cu2+ systems.
| System | 3j (mmol/L) | 3a (mmol/L) | NaOH (mol/L) | H2O2 (mol/L) |
| 3j-H2O2-Cu2+ | 0.1 | – | 0.25 | 0.01 |
| 3a-H2O2-Cu2+ | – | 0.1 | 0.25 | 0.005 |
Moreover, the application of the other compounds synthesized in the CL field is studied in our analytical chemistry laboratory.
4 Conclusions
In conclusion, we have developed a novel and efficient method for the synthesis of trisubstituted imidazole derivatives starting from solid-phase raw material. Microwave-assisted, solvent-free, solid support, as a green chemistry procedure, could be applied to the synthesis of heterocyclic compounds including nitrogen atom. The method afforded the guidance to synthesize substituted imidazole derivatives from the α-carboxyl esters. The synthesis of other trisubstituted imidazoles containing benzene rings or else using the method is currently ongoing in our laboratory. Besides, the synthesized compounds will possess wide application potential to chemical testing.