

1 Introduction
Catharanthus roseus (Madagascar periwinkle) is a perennial herb or ornamental of the Apocynaceae producing over 130 terpenoid indole alkaloids (TIAs) of which the metabolic compounds vinblastine and vincristine are used as anticancer agents [1]. These two compounds are synthesized in the indole alkaloid biosynthesis pathway. C. roseus also harbours a large number of phenolic compounds via the phenylpropanoid biosynthesis pathway [2–4] that induce several defence mechanisms against biotic and abiotic stresses [5,6].
Large efforts have been made for improving the biosynthesis of the two anticancer compounds of Catharanthus roseus (L.) G. Don. However, these efforts are yet to be potentially successful at the commercial-scale in reducing the cost of the therapy. Therefore, it is a demand to detect the mechanisms of metabolic regulation by which the indole alkaloids biosynthesis pathway takes the priority over other competing mechanisms. Elicitating core enzymes in the TIA pathway and acquiring more information on this obscure pathway likely raise the chance to enhance the production of several other valuable compounds. Alkaloids in the pathway such as ajmalicine and serpentine are commercially used against hypertension [1] and circulatory disorders [7]. The two bisindoles vinblastine and vincristine are derived from coupling vindoline and catharanthine in the TIA pathway [8]. The pathway involves three main stages towards the biosynthesis of the two bisindole alkaloids. The first is the formation of tryptamine and secologanin from the shikimate and monoterpenoid biosynthesis pathways, respectively. The first compound is driven by tryptophan synthase, while the second is driven by secologanin synthase. The second stage includes the core reaction in the TIA pathway, which is the formation of monomeric alkaloid strictosidine that is eventually converted to either vindoline or catharanthine [9]. Formation of strictosidine requires the action of strictosidine synthase [10]. Towards the formation of vindoline from tabersonine, six steps are required, of which five were deciphered up to date. They involve the enzymes tabersonine 16-hydroxylase, O-methyltransferase, N-methyltransferase, desacetoxyvindoline-4-hydroxylase and deacetylvindoline-4-O-acetyltransferase [11]. The third stage is the formation of the two bisindole alkaloids after the eventual condensation of catharanthine and vindoline [12,13]. Compound produced instantly from the condensation is α-3′,4′-anhydrovinblastine, which is converted into vinblastine that is further converted into vincristine. The vacuolar class-III peroxidase acts directly upstream the step of biosynthesis of the two bisindoles in Catharanthus roseus [8].
Methyl jasmonate (MeJA) was reported to elicit secondary metabolites in plant, such as alkaloids and phenolic compounds, to regulate plant growth and adaptation [5,14–17]. Rijhwani and Shanks [18] indicated that MeJA increases the yield of alkaloids of the TIA pathway in C. roseus hairy root cultures, while Cocetta et al. [16] indicated that MeJA modulates phenolic metabolism in blueberry. More recently, Liu et al. [19] studied the combinational effects of ethylene and MeJA on indole alkaloid and phenolic compounds profiles in C. roseus and provided new insights on metabolisms governing the production of the two types of compounds in leaves of C. roseus. Jasmonic acid (JA) was also reported to play a major role in the biosynthesis of toxic secondary metabolites [19] and as an elicitor towards enhancing several steps in the indole alkaloid pathway [20].
In the present study, we aim to detect the effects of MeJA (6 uM) treatment across time, e.g., 2, 6, 12 and 24 days, in a trial to detect the stepwise response of MeJA-induced genes and pathways in leaves of C. rouses.
2 Methods
2.1 Materials
Seeds of C. roseus were left to germinate in the greenhouse of King Abdulaziz University, Jeddah, KSA complying with the institutional, national and international guidelines. No ethics approval was required for any aspect of this study. Plantlets were grown in pots containing sterilized soil: vermiculate at 1:1 and watered with sterile ddH2O for 4 weeks. Seedlings were grown at 25 °C with 16-h light periods, ∼125 uE·m−2·s−1 light intensity and ∼70% humidity. Evaporated water was replaced daily. Then, 4-week-old seedlings were watered with MeJA (6 uM) and leaf samples were collected after 0, 6, 12, and 24 days. Concurrently, control seedlings were watered with the solvent DMSO at equivalent volume. Leaf samples of the two growth conditions were collected in two replicates at the different time points.
2.2 RNA-Seq analysis
Flash-frozen similar-sized leaf samples were crushed into a fine powder in microcentrifuge tubes and total RNAs were extracted, quantified and checked for presence of DNA contamination as previously described [21]. Then, RNAs were shipped to BGI, China, for deep sequencing and raw data were submitted to the National Centre for Biotechnology Information (NCBI) (submission ID: SUB2671065, BioProject ID: PRJNA386850). Filtered reads were de novo assembled and quantified as previously described [22]. Raw reads were mapped to the recovered de novo assembled contigs (transcripts). Differentially expressed (DE) transcripts were annotated and KEGG pathway analyses were performed using blast-2-GO software (version 2.3.5, http://www.blast2go.com/) and GO terms were obtained with the default parameters. Clusters with functional enrichment were identified via significant Pearson correlation and permutation analysis [23]. Subsequent clustering was done as previously described [22]. RPKM-derived read counts (RPKM ≥ 2) were compared using a likelihood ratio test [24]. To determine DE transcripts, ≥ 2-fold change in expression and false discovery rate (FDR) of ≤ 10−3 was used. Resulted data were subjected to hierarchical clustering using the Pearson correlation as the distance metric.
2.3 Semi-quantitative RT-PCR
RNA-Seq data was further validated via semi-quantitative RT-PCR for transcripts upregulated at 6-day-time point. The actin gene of C. roseus was used as the house-keeping gene. Primers (Table S1) were designed using the Netprimer software (http://www.premierbiosoft.com/netprimer/index.html) with the criteria previously described [25].
3 Results and discussion
3.1 Statistics of the sequencing datasets
Sequencing of RNA samples of C. roseus leaves under the two growth conditions across time yielded a total of ∼255 million reads corresponding to an average of ∼21 billion nucleotides of cDNA per sample (Table 1). Around 72% of the total reads mapped to the de novo assembled contigs, ∼60% of the total reads showed perfect matches, while ∼12% showed mismatches. Around 45 and 27% of the total reads showed unique and multi-position matches, respectively. Around 28% of the total reads unmapped to the de novo assembled contigs. This indicates that the unmapped reads were not incorporated in the originally de novo assembled contigs.
Statistics RNA-Seq numerical data analysis of C. roseus leaves.
| Sample ID | Total No. readsa | No. mapped readsb | (%)c | No. reads with perfect match (%)d | No. reads with mismatches (%)e | No. of reads with unique matches (%)f | No. reads with multi-position matches (%)g | No. unmapped reads (%)h |
| C-W (1) | 24,122,605 | 17,469,363 | 72.42 | 14,696064 (60.9) | 2,773,299 (11.5) | 10,330,698 (42.8) | 7,138,665 (29.5) | 6,653,241 (27.6) |
| C-W (2) | 24,120,444 | 17,589,969 | 72.93 | 14,481605 (60.0) | 3,108,364 (12.8) | 10,361,874 (43.0) | 7,228,095 (29.9) | 6,530,474 (27.1) |
| 6D-W | 24,121,216 | 18,088,051 | 74.99 | 15,201880 (63.0) | 2,886,171 (11.9) | 11,299,801 (46.8) | 6,788,250 (28.1) | 6,033,164 (25.0) |
| 12D-W (1) | 15,023,698 | 10,996,216 | 73.19 | 9,176,725 (61.08) | 1,819,491 (12.1) | 6,893,581 (45.9) | 4,102,635 (27.3) | 4,027,481 (26.8) |
| 12D-W (2) | 24,117,952 | 17,799,779 | 73.80 | 14,772,072 (61.2) | 3,027,707 (12.5) | 11,246,111 (46.6) | 6,553,668 (27.2) | 6,318,172 (26.2) |
| 24D-W (1) | 24,110,564 | 16,642,629 | 69.03 | 13,733,007 (57.0) | 2,909,622 (12.0) | 10,224,138 (42.4) | 6,418,491 (26.6) | 7,467,934 (31.0) |
| 24D-W (2) | 24,120,004 | 16,917,271 | 70.14 | 13,990,560 (58.0) | 2,926,711 (12.1) | 10,449,089 (43.3) | 6,468,182 (26.8) | 7,202,732 (29.9) |
| 6D-MJ | 23,230,225 | 16,690,402 | 71.85 | 14,099,088 (60.7) | 2,591,314 (11.1) | 10,020,620 (43.1) | 6,669,782 (28.7) | 6,539,822 (28.2) |
| 12D-MJ (1) | 24,117,745 | 17,822,229 | 73.90 | 14,653,856 (60.8) | 3,168,373 (13.1) | 11,409,242 (47.3) | 6,412,987 (26.5) | 6,295,515 (26.1) |
| 12D-MJ (2) | 24,121,278 | 17,797,991 | 73.79 | 14,979,278 (62.1) | 2,818,713 (11.7) | 11,438,173 (47.4) | 6,359,818 (26.3) | 6,323,286 (26.2) |
| 24D-MJ (1) | 24,076,941 | 17,238,193 | 71.60 | 14,452,921 (60.0) | 2,785,272 (11.5) | 10,690,263 (44.4) | 6,547,930 (27.2) | 6,838,747 (28.4) |
| 24D-MJ (2) | 24,040,120 | 16,990,589 | 70.68 | 14,249,950 (59.2) | 2,740,639 (11.4) | 10,459,786 (43.5) | 6,530,803 (27.1) | 7,049,530 (29.3) |
a Total number of reads recovered from RNA-Seq.
b Number of mapped reads to the de novo assembled contigs.
c Percentage of mapped reads over total No. of reads.
d Number and percentage of reads with perfect match over total No. of reads.
e Number and percentage of mismatch reads over total No. of reads.
f Number and percentage of reads with unique match over total No. of reads.
g Number and percentage of reads with multi-position match over total No. of reads.
h Number and percentage of reads unmapped to the assembled contigs over total No. of reads.
3.2 Cluster analysis of DE transcripts due to MeJA treatment
Globally, a number of 9456 DE transcripts was recovered (Table S2) and distributed in 294 expression profiles or clusters (Fig. S1). Hierarchical cluster analysis of gene expression under either condition across different time points of MeJA treatment indicated the high quality of sampling and RNA-Seq analysis as replicates of each treatment at a given time point were clustered together (Fig. 1). Two-dimensional hierarchical clustering classified DE profiles according to the similarity of their expression profiles of which three (Nos. 13, 142 and 216) and one (No. 165) clusters fitted within the required expression profiles of upregulation after 6 and 12 days of MeJA treatment, respectively (Fig. 2 and Table S2). There is only one cluster (No. 12) that showed upregulation after both 12 and 24 days of MeJA treatment and one cluster (No. 79) for upregulation after 24 days of MeJA treatment (Table S2 and Fig. S1). A number of 11 clusters (No. 5, 26, 33, 62, 67, 71, 121, 130, 154, 182, and 284) showed upregulation after MeJA treatment across all time points. Semi-quantitative RT-PCR (sqRT-PCR) of six upregulated transcripts at the TIA pathway was performed to validate the RNA-Seq data (Fig. S2). The analysis of sqRT-PCR indicated that the selected genes were upregulated only after 6 days of MeJA treatment supporting the results of RNA-Seq analysis.
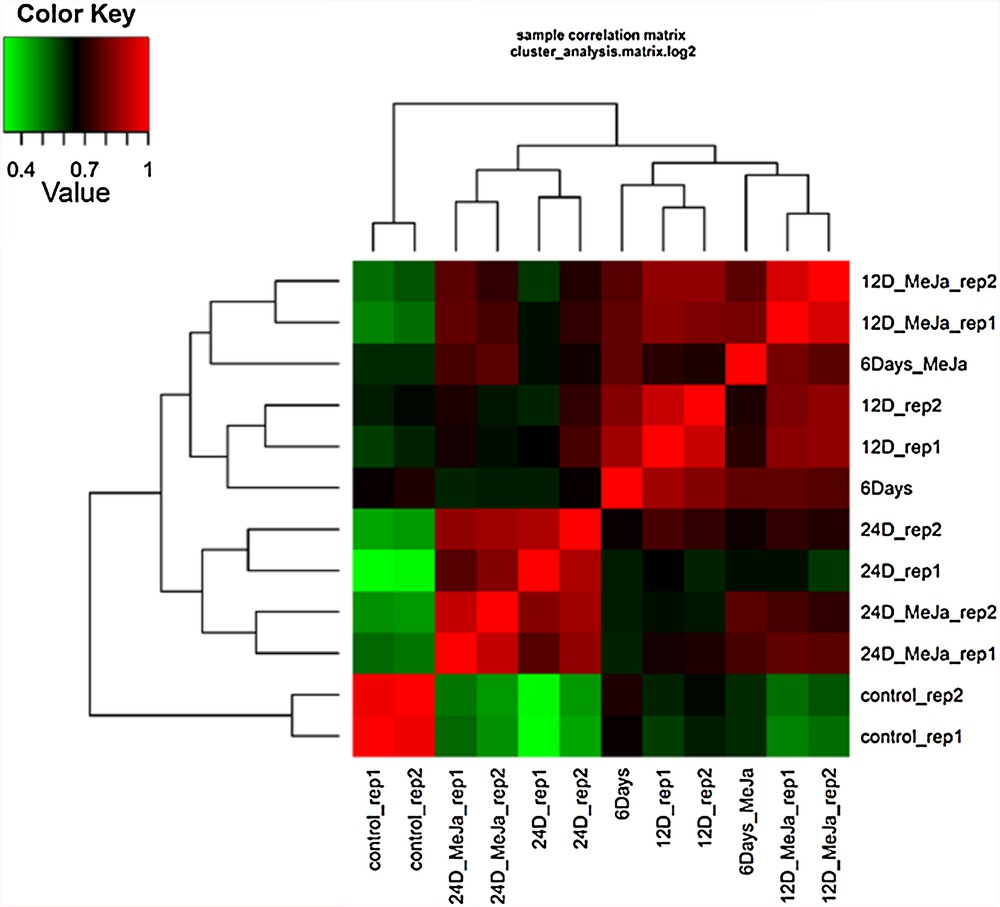
Hierarchical cluster analysis of gene expression based on log ratio RPKM data for C. roseus leaf transcriptome under water and MeJA (6 uM) treatment for 0, 6, 12 and 24 days. Rate of gene expression is referred to by a 0 (green) to 1 (red) scale. C.
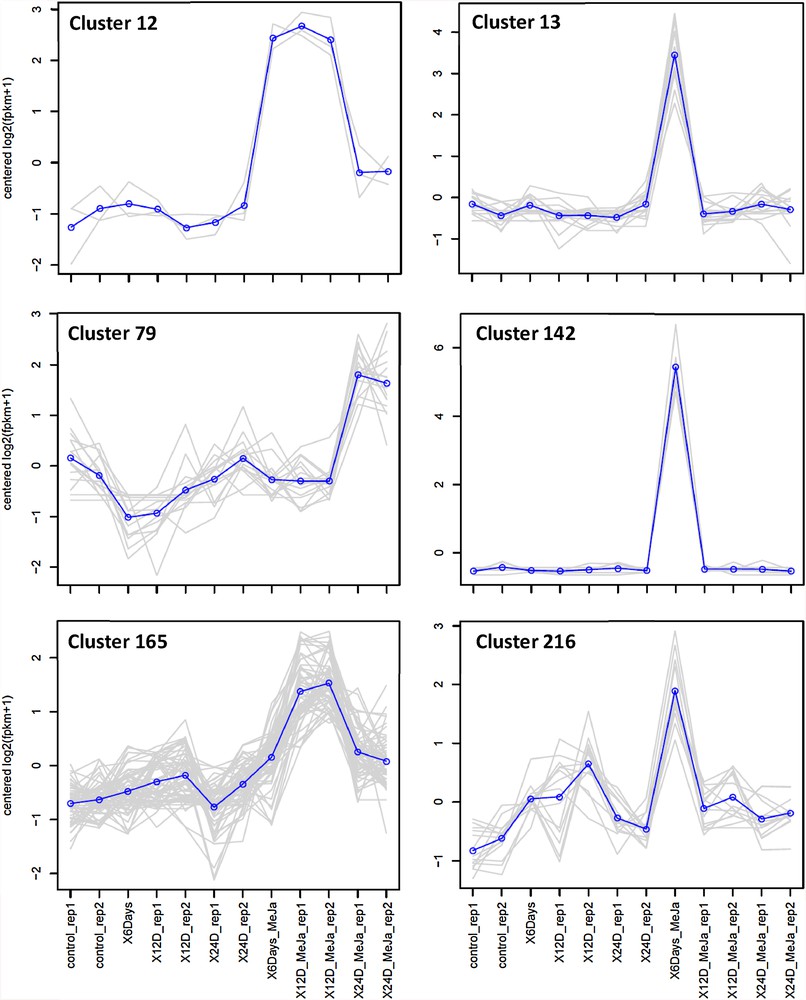
Selected clusters of upregulated genes of C. roseus leaves after 6 (clusters 13, 142 and 216), 12 (cluster 165), 24 (cluster 79) h and both 6 and 12 days (cluster 12) of MeJA (6 uM) treatment. Blue lines indicate overall expression pattern across different transcripts of a given cluster.
The most relevant upregulated genes in terms of their involvement in the TIA and upstream pathways as well as in those related to phenolic compound biosynthesis were further analysed. The regulated pathways after 6 days of MeJA treatment are “indole alkaloid biosynthesis or TIA”, “phenylalanine, tyrosine and tryptophan biosynthesis or PTT” and “monoterpenoid biosynthesis or MT” (Fig. 3 and S3–S4, respectively). While, the major regulated pathways after 12 days of MeJA treatment are “phenylpropanoid biosynthesis or PP”, “anthocyanin biosynthesis or AC” and “ubiquinone and other terpenoid-quinone biosynthesis or UTQ” (Figs. S5–S7, respectively).
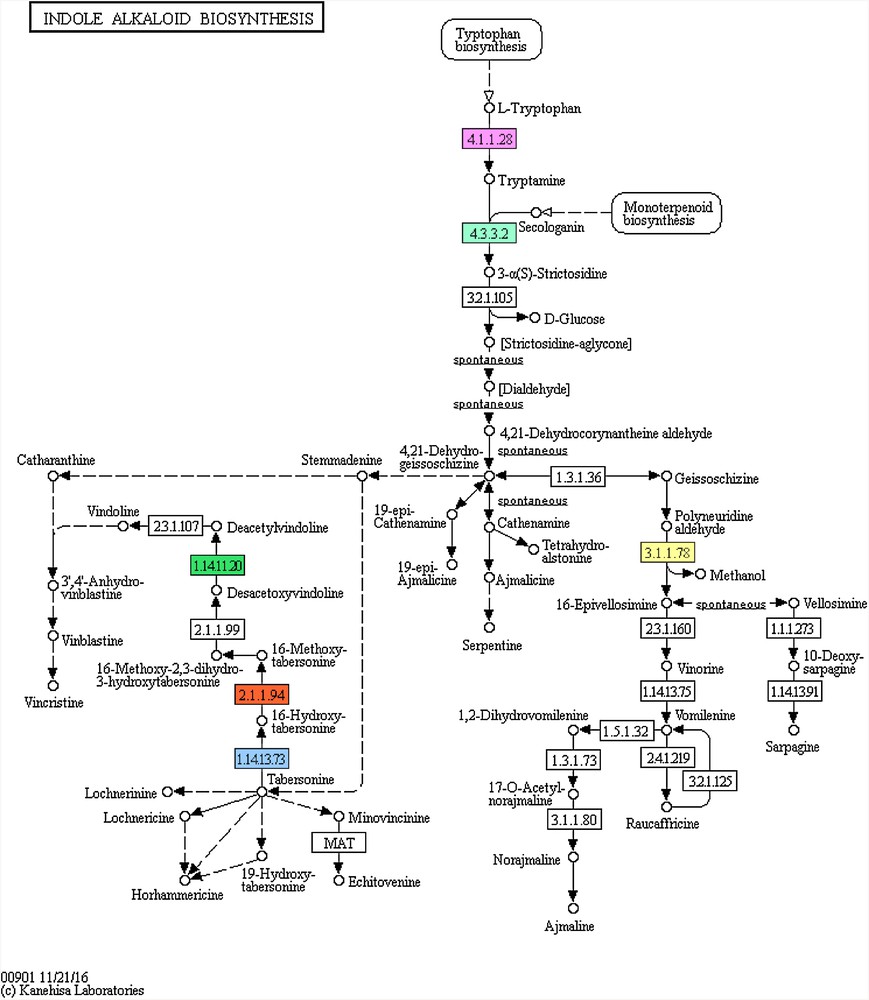
Enzymes in the “indole alkaloid biosynthesis or TIA” pathway in C. roseus leaves activated under MeJA (6 uM) treatment for 6 days. Activated enzymes are shown in coloured boxes, while the enzymes with unchanged activation rates are shown in uncoloured boxes. Different box colours in the pathway indicates different activated enzymes. EC: 1.14.13.73 = tabersonine 16-hydroxylase or T16H (blue box), EC: 2.1.1.94 = tabersonine 16-O-methyltransferase or OMT (red box), EC: 1.14.11.20 = deacetoxyvindoline-4-hydroxylase or D4H (green box), EC: 4.1.1.28 = tryptophan decarboxylase or TDC (violate box), EC: 4.3.3.2 = strictosidine synthase or STR (light green box), EC: 3.1.1.78 = polyneuridine-aldehyde esterase or PNAE (yellow box).
Six enzymes encoded by genes in the TIA pathway were activated after 6 days of MeJA treatment (Fig. 3 and Table S2). Three of these genes encode tabersonine 16-hydroxylase (EC: 1.14.13.73, T16H, clusters 13 and 216), tabersonine 16-O-methyltransferase (EC: 2.1.1.94, OMT, cluster 216) and deacetoxyvindoline-4-hydroxylase (EC: 1.14.11.20, D4H, clusters 13 and 216) towards the biosynthesis of vindoline. Two other genes encode enzymes in two consecutive steps in the TIA pathway. These enzymes are tryptophan decarboxylase (EC: 4.1.1.28, TDC, cluster 216) and strictosidine synthase (EC: 4.3.3.2, STR, cluster 142) that altogether contribute to the conversion of tryptophan to strictosidine (Fig. 3). Otherwise, this amino acid likely act as a substrate for two other pathways (Fig. 4). The first is “tryptophan metabolism” pathway of which tryptophan is depleted towards the production of indole acetic acid, which in turn contribute to plant growth and hormone signal transduction pathway. The other pathway is “staurosporine biosynthesis” towards the production of several valuable drugs. Several routes in the TIA pathway can be followed due to the action of the core gene TDC such as biosyntheses of catharanthine, ajmalicine, serpentine and/or ajmaline. Interestingly, the sixth regulated gene, encoding polyneuridine-aldehyde esterase (EC: 3.1.1.78, PNAE, cluster 13), acts in other route in the TIA pathway towards the possible production of ajmaline, raucaffricine, and/or sarpagine.
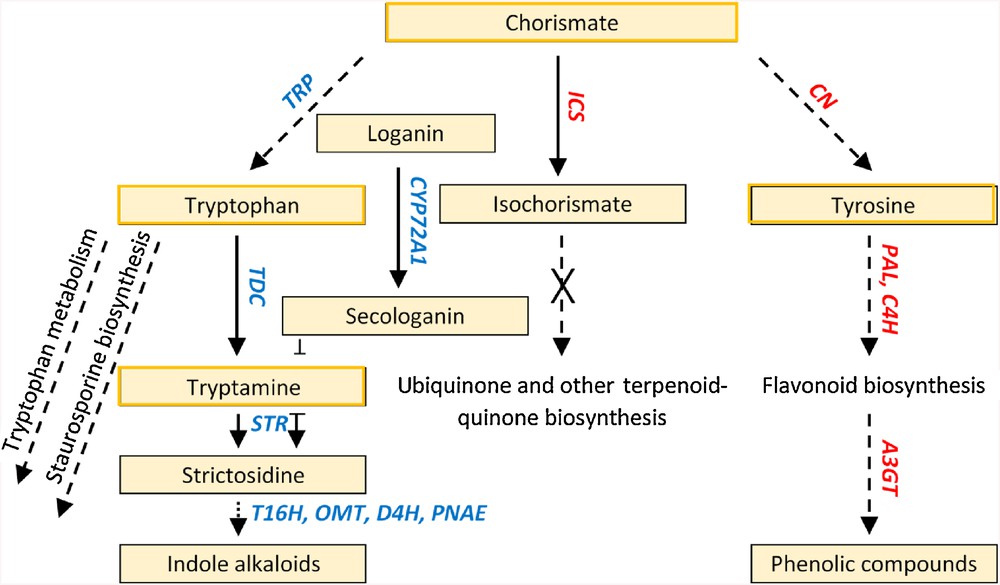
Activated steps in the pathways of “indole alkaloid biosynthesis”, “phenylalanine, tyrosine and tryptophan biosynthesis”, “monoterpenoid biosynthesis”, “phenylpropanoid biosynthesis”, “anthocyanin biosynthesis” and “ubiquinone and other terpenoid-quinone biosynthesis” due to MeJA treatment. Genes indicated in blue are upregulated after 6 days of treatment, while those indicated in red are upregulated after 12 days of the treatment. Boxes for compounds act upstream master switches are orange-lined. Solid arrows indicate single-step reactions and dashed arrows indicate multi-step reactions. “X” on arrow indicates the unlikely occurrence of this step in the pathway.
Genes encoding two other enzymes acting upstream of the six enzymes of the TIA pathway were also regulated after 6 h of MeJA treatment. They are tryptophan synthase (EC: 4.2.1.20, TRP, clusters 13 and 142) and secologanin synthase (EC: 1.3.3.9, CYP72A1, cluster 13) in the PTT (Fig. S3) and MT (Fig. S4) pathways, respectively. There are several other enzymes in the MT pathway that were activated due to MeJA treatment but none of them contributes in the promotion of TIA pathway.
The other three pathways are PP (Fig. S5), AC (Fig. S6) and UTQ (Fig. S7). The first two pathways mostly contribute to the biosynthesis of phenolic compounds, while the third is not normally activated in plant. A pathway that connect PP and AC pathways is the flavonoid biosynthesis (FN) pathway. Interestingly, the FN pathway is not regulated under MeJA treatment, but it looks that this negative response made no barrier to the flow of information from PP to AC pathways. The main concern towards the activation of enzymes in the pathways contributing to the phenolic compound biosynthesis is that they might compete with enzymes in the TIA, PTT and MT pathways that contribute to the indole alkaloid biosynthesis. This concern is high in case the enzymes of the two sets of crosstalking pathways are concurrently activated under MeJA treatment.
The PP pathway that starts with the conversion of phenylalanine to cinnamic acid by phenylalanine ammonia lyase (EC: 4.3.1.24, PAL, cluster 165) and the downstream AC pathway (Fig. S6) that starts with the conversion of pelargonidin to several phenolic compounds by anthocyanidin 3-O-glucosyltransferase (EC: 2.4.1.115, A3GT, cluster 165) compete with the TIA pathway for the precursor chorismate [4]. The latter enzyme was triggered in the present study by MeJA treatment at all time points (Fig. S1 and Table S2). In addition, the activation of chorismate mutase (EC: 5.4.99.5, CM, cluster 165) of the PTT pathway (Fig. S8) and isochorismate synthase (EC: 5.4.4.2, ICS, cluster 165) of the UTQ pathway (Fig. S7) after 12 h of MeJA treatment promote the latter two pathways. No explanation for the latter pathway being activated as this is unlikely in plant. However, we speculate that earlier activation (e.g., 6 h after MeJA treatment) of tryptophan synthase (cluster 13, Fig. S1) likely makes TIA pathway a more favourable target as this enzyme as well as secologanin synthase might act as a master switch towards the promotion of the TIA pathway.
Anthocyanins are synthesized due to the action of the gene encoding anthocyanidin 3-O-glucosyltransferase (a3gt). The enzyme acts downstream of the FN biosynthesis pathway. The latter pathway occurs downstream the PP biosynthesis pathway. The enzyme that triggers these consecutive reactions is phenylalanine ammonia-lyase (PAL) of the PP biosynthesis pathway. Then, we speculate that PAL gene acts as the master switch of several gene expression in the leaves treated with MeJA in the PP pathway and the downstream FN, then, AC pathways towards the production of several phenolic compounds. Flow of information starts with triggering expression of the gene (c4h) encoding trans-cinnamate 4-monooxygenase (EC: 1.14.13.11, C4H, cluster 165) in the PP biosynthesis pathway and ends with the expression of anthocyanidin 3-O-glucosyltransferase (a3gt) in the AC biosynthesis pathway. This type of crosstalking of pathways has been argued in earlier reports [4]. MeJA was reported earlier to accelerate fruit ripening in Fragaria by increasing soluble solid content and anthocyanin accumulation [26]. This indicates that the PP and AC pathways are favoured at later stages of plant development.
Light is thought to significantly influence the biosynthesis of vindoline of which three genes, e.g., tdc, d4h and dat were proven to be light-inducible [27]. The first two genes were upregulated in the present study after 6 days of MeJA treatment, while the third was not. The two regulated genes might harbour elements upstream the promoter sequences that positively respond to either light or MeJA signals, or otherwise, light might induce biosynthesis of indigenous MeJA in the plant cells. The latter speculation indicates that light influences the biosynthesis of vindoline indirectly. Interestingly, one light-responsive enzyme namely zeaxanthin epoxidase (EC: 1.14.15.21), in the carotenoid biosynthesis (or CT) pathway for the production of the photoprotective violaxanthin [28], was activated (Fig. S9) after 12 days of MeJA treatment (cluster 165). Under high light condition, violaxanthin deepoxidates to zeaxanthin to facilitate safe removal of excess excitation energy. Additionally, a gene encoding short-chain alcohol dehydrogenase basically acts in MeJA biosynthesis [29]. This gene is upregulated after 12 days of MeJA treatment (cluster 165). This indicates that MeJA might stimulate the expression of the genes synthesizing it under harsh condition. In turn, this action promotes ABA biosynthesis in plants leading to reduced grain yield.
3.3 bHLH transcription factors (TFs) triggered by MeJA treatment
The basic helix-loop-helix (bHLH) represents one of the largest families of dimerizing transcription factors [30]. In the present study, two TFs were regulated after 6 days of MeJA treatment (Fig. 5). Of which, one (e.g., bHLH25) was upregulated (cluster 13), while the other (e.g., bHLH14) was downregulated (cluster 224). Cluster analysis indicated the coexpression of bHLH25 and STR genes (clusters 13 and 142, respectively; Fig. 5) in alignment with our previous findings [22] of which knockdown of the TF proved its role in driving STR gene via virus-induced gene silencing (VIGS). bHLH25 is mainly expressed in flower and silique of Arabidopsis under MeJA treatment [22,31]. The previous results also indicated coexpression of t16h gene with three TFs, two of which harbour the B3 domain, while the third is a bHLH135. Although t16h gene was upregulated after 6 days of MeJA treatment, none of the three TFs are regulated under MeJA treatment. This indicates that t16h gene might either be triggered by the action of one or more TFs or by MeJA treatment, while the TF genes are not definitely triggered by MeJA treatment. Otherwise, coexpression of the TFs and t16h genes is arbitrary. The other bHLH, e.g., bHLH14 is part of the bHLH subgroup IIId TFs acting as transcription repressors of jasmonic acid (JA) responses, such as the induction of anthocyanin accumulation as well as the block of plant defence against pathogens and insects [32]. This transcription repressor competes with the transcription activators, e.g., MYC2/MYC3/MYC4 and WD-repeat/bHLH/MYB complex, in binding their target sequences. Thus, downregulation of bHLH14 results in the induction of MeJA towards the accumulation of anthocyanin in accordance with our results. The coordinated regulation of JA responses acts as an attenuator of plant defence and development as well as JA-induced leaf senescence [33] that is induced to relocate nutrients and energy from old leaves to newly developing leaves and organs to eventually promote plant growth, reproduction and defence.
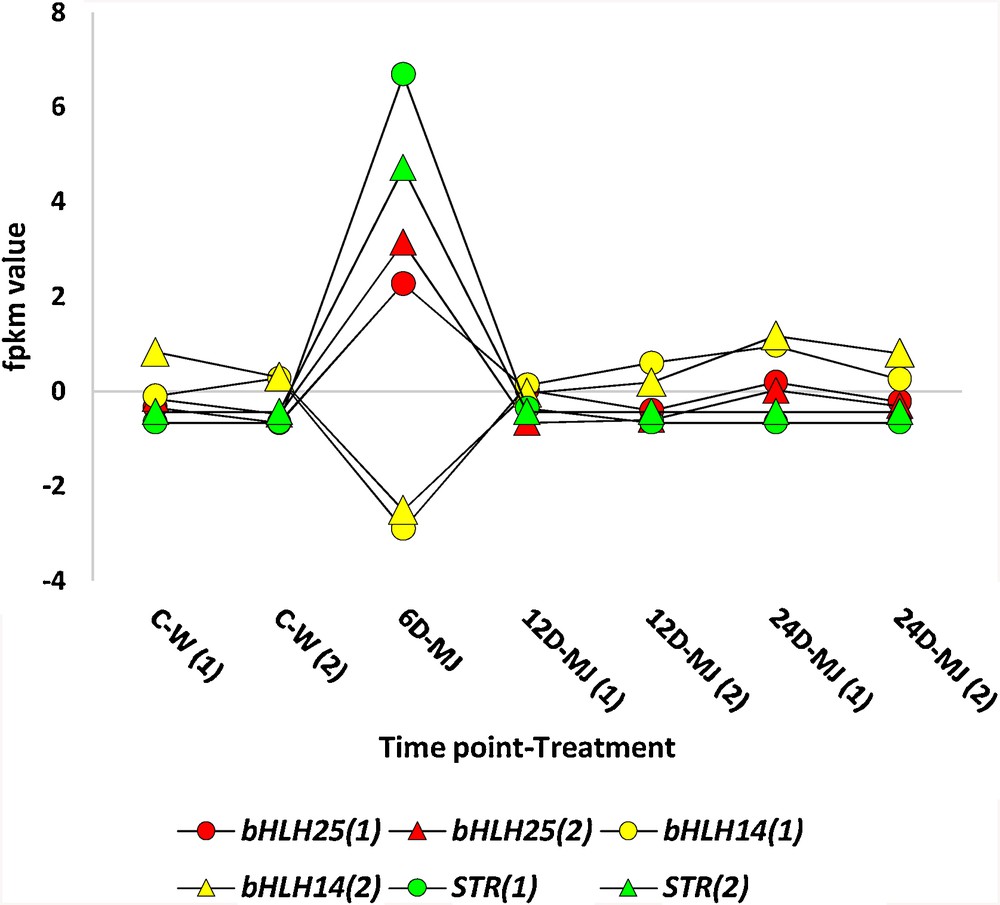
Regulation of STR (1 & 2), bHLH25 (1 & 2) and bHLH14 (1 & 2) transcripts of clusters 142, 13 and 224, respectively, at 6-d time point of MeJA (6 uM) treatment in C. roseus leaves. Two versions of transcripts STR (green round and rectangle shapes) as well as bHLH25 (red round and rectangle shapes), on the one hand, and two other versions of bHLH14 (yellow round and rectangle shapes), on the other hand, were upregulated and downregulated, respectively. Values of fpkm are shown in Table S2. The figure indicates MeJA treatment time points of 0 (C–W), 6, 12 and 24 days, only.
In earlier reports, auxins were reported to downregulate the expression of the genes encoding core enzymes in the bisindole alkaloid biosynthesis in C. roseus [34] such as tdc, while cytokinins (CKs) are proven to upregulate this gene [35]. Interestingly, treatment with exogenous MeJA stimulates the production of CKs, thus might indirectly upregulate tdc gene in C. roseus [36]. We speculate that the aforementioned stimulation might be triggered by MeJA-induced CK rather than being directly triggered by MeJA. Several genes in the TIA pathway responded differently to MeJA treatment in terms of expression rate and timing of which d4h gene followed by STR then TDC genes showed the strongest response [37,38]. Vázquez-Flota et al. [39] also indicated that MeJA induces the biosynthesis of several alkaloids in the TIA pathway in C. roseus such as ajmaline, ajmalicine, vindoline and catharanthine. Other reports indicated that the biosyntheses of catharanthine, vindoline, and vinblastine in C. roseus cultured cells were accompanied by upregulation of tdc, t16h, and d4h [40,41]. Based on the results of the present study with regard to the latter three genes, MeJA was proven to contribute to the biosynthesis of the three important indole alkaloids catharanthine, vindoline, and vinblastine of the TIA pathway.
4 Conclusion
The present work aimed at detecting the prolonged effects of MeJA across time (up to 24 days) in order to detect the stepwise response of MeJA-induced genes and pathways in leaves of C. rouses. The genes encoding TDC and STR might concurrently act as a master switch of the TIA pathway towards the production of the indole alkaloids. While the gene encoding PAL enzyme might act as the master switch of phenylpropanoid biosynthesis and the downstream flavonoid biosynthesis and anthocyanin biosynthesis pathways towards the production of several phenolic compounds. Concordant activities of the flower- and silique-specific bHLH25 transcription factor and the key enzyme in the TIA biosynthesis pathway namely STR were suggested. We speculate that enzymes activated earlier might make TIA biosynthesis pathway more favourable in C. roseus than anthocyanin biosynthesis pathway. Transcriptomic data of the present study can provide resources for future improvement of this important medicinal wild plant via metabolic engineering towards the overproduction of the two important basindoles at the commercial scale.
Authors’ contributions
All authors have read and approved this manuscript. Conceived and designed the experiments: AB AA SE NOG MA AMR MGA FE. Performed the experiments: AA AA MAA SB JSMS. Analyzed the data: AA HSA FME. Wrote the paper: AB SB MGA JSMS MEN FME.
Disclosure of interest
The authors declare that they have no competing interest.
Acknowledgements
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, the Kingdom of Saudi Arabia–award No. 12-BIO3064-03. The authors also acknowledge with thanks Science and Technology Unit, King Abdulaziz University, for technical support.