

1 Introduction
Cetartiodactyla is one of the most diversified orders of mammals, with 332 extant species grouped into 132 genera [1]. Members of this taxonomic group were originally divided into two different orders: Artiodactyla and Cetacea [2].
Artiodactyla contains 247 species of hoofed mammals, which are currently grouped into 10 families [1]: (1) Bovidae (the most diversified family with 142 species related to cattle, goat and sheep), (2) Cervidae (deer, muntjac, moose, etc.; 54 species), (3) Moschidae (musk deer; 7 species), (4) Giraffidae (giraffe and okapi), (5) Antilocapridae (pronghorn; 1 species), (6) Tragulidae (chevrotains; 10 species), (7) Suidae (pigs; 18 species), (8) Tayassuidae (Peccaries; 4 species), (9) Hippopotamidae (hippos, 2 species), and (10) Camelidae (camel, dromedary, llama and vicugna; 7 species) [1]. Artiodactyls are characterized by two main limb features: (i) a paraxonic foot structure in which the axis of the limb support passes between the third and fourth digits, which carries most of the animal's weight; and (ii) in the ankle where the astragalus is ‘double-pulleyed’, i.e. with a deep trochlea for the tibia and an opposing trochlea for the navicular, which enhances hind limb flexion and extension and allows very limited lateral rotation of the foot. Artiodactyls were originally present on all continents, except the Australian region and Antarctica. Most domestic livestock come from this group, including cattle, buffalo, sheep, goat, pig, and camel.
Cetacea contains 14 species of baleen whales (Mysticeti; 4 families: Balaenidae, Balaenopteridae, Eschrichtiidae, and Neobalaenidae) and 71 species of toothed whales, which include dolphins, porpoises, sperm whales, beaked whales, and others (Odontoceti; 8 families: Delphinidae, Lipotidae, Monodontidae, Phocoenidae, Physeteridae, Platanistidae, Iniidae and Ziphiidae) [1]. All are marine animals except a few species of freshwater dolphins (e.g., Inia geoffrensis, Lipotes vexillifer, Platanista gangetica, Sotalia fluviatilis, Orcaella brevirostris) [3]. The common ancestor of Cetacea acquired many adaptations to an aquatic life, such as a fusiform body, forelimbs modified into flippers, no hindlimbs or rudiments, and a tail fin (fluke) used for propulsion [4,5].
Molecular studies have concluded to the paraphyly of Artiodactyla, due to the sister–group relationship between Cetacea and Hippopotamidae [6–8]. To render taxonomy compatible with this new phylogeny, Montgelard et al. [6] proposed to place all species of Artiodactyla and Cetacea into the same order, called Cetartiodactyla. The two main limb features found in all extant terrestrial cetartiodactyls, i.e., a double-pulley astragalus and a paraxonic foot, have been identified in Eocene whales [9], indicating that these two key adaptations occurred in the common ancestor of Cetartiodactyla.
Both morphological and molecular studies have supported the monophyly of six cetartiodactyl higher taxa (above the family level): (1) Cetacea; (2) Mysticeti; (3) Odontoceti [10,11]; (4) Suina, which unites the families Suidae and Tayassuidae [6]; (5) Pecora, which groups the five families Antilocapridae, Bovidae, Cervidae, Giraffidae, and Moschidae; and (6) Ruminantia, which includes Pecora and Tragulidae [12]. In addition, molecular studies have revealed that the Cetacea is the sister–group to the Hippopotamidae, followed by the Ruminantia [8,11,13]. The association of Cetacea and Hippopotamidae, termed the Whippomorpha [14], has been also corroborated by morphological analyses [15]. By contrast, no morphological support has yet been found for the association of Whippomorpha and Ruminantia, a clade termed the Cetruminantia [14]. Molecular analyses have however produced conflicting signals for basal relationships within Cetartiodactyla: Arnason et al. [16] suggested a sister–group relationship between Suina and Camelidae; Matthee et al. [8] found an early divergence of Suina; whereas Murphy et al. [17] and Zhou et al. [11] concluded to an early divergence of Tylopoda. Deep relationships within Odontoceti, Pecora, and Bovidae have been poorly resolved, suggesting the occurrence of rapid radiation events [12,18–20].
Most previous studies dealing with the molecular systematics of Cetartiodactyla were based on reduced taxon sampling (e.g., 12 species in Gatesy et al. [7]; 34 species in Matthee et al. [8]; 21 species in Zhou et al. [11]), small amount of phylogenetic information, i.e., only the cytochrome b gene [21], or supermatrices including large amount of missing data (e.g., 62% in O’Leary and Gatesy [13]). As a consequence, no comprehensive time-calibrated tree is available for analyzing the evolution of Cetartiodactyla as a whole. In this study, we analyzed a dataset including 210 taxa for an alignment of the complete mitochondrial genome (14,902 nucleotide characters). Our analyses include all families, 81% of the genera and 55% of the species currently described in the order Cetartiodactyla [1]. For several species, such as Dorcatragus megalotis, Madoqua saltiana, and Neotragus batesi, we present here the first molecular data ever collected. This large dataset was analyzed to better understand how cetartiodactyls evolved during the Cenozoic era, with a particular interest in the identification of rapid radiations in the suborder Ruminantia. This suborder is indeed particularly interesting because ruminants are widely represented in the fossil record of the Neogene. Today, the suborder Ruminantia includes 59% of the genera and 65% of the species currently described within Cetartiodactyla [1]. Paradoxically, ruminants were poorly represented in previous molecular studies. Here, we made a strong genome sequencing effort to cover a large spectrum of species.
The use of complete mtDNA genomes and the absence of missing data guarantee sufficient amount of phylogenetic information at different taxonomic levels, including family, subfamily, tribe, genus and species. Our three major objectives were: (i) to produce a reliable tree for interpreting phylogenetic relationships among cetartiodactyls; (ii) to determine the major phases of diversification; and (iii) to correlate some of these episodes with climatic, biogeographic, and ecological events by using a chronogram inferred from our molecular data.
2 Materials and methods
2.1 DNA extraction, amplification and sequencing
The complete mitochondrial genome was amplified and sequenced for 128 taxa, all but two (the water buffalo B. bubalis and camel C. dromedarius) collected from the wild (Appendix 1). Total DNA was extracted from blood, hair, muscle, skin, or cryopreserved cells as detailed in Ropiquet and Hassanin [22]. At least twenty-three overlapping fragments of the mitochondrial genome were amplified by polymerase chain reaction (PCR) using published primers [23], as well as new primers (for which sequences are detailed in Appendix 2). Amplifications were done in 30 μl using 3 μl of Buffer 10 X with MgCl2, 3 μl of dNTP (6.6 mM), 0.15 μl of Taq DNA polymerase (2.5 U), and 1.5 μl of the two primers at 10 μM. The standard PCR conditions were as follows: 3 min at 94 °C; 35 cycles of denaturation/annealing/extension with 1 min at 94 °C for denaturation, 1 min at 50–60 °C for annealing, and 1 min at 72 °C for extension; and 7 min at 72 °C. Both strands of all PCR products were sequenced using Sanger sequencing on an ABI 3730 automatic sequencer at the Centre National de Séquençage (Genoscope) in Evry (France). The sequences were analyzed and assembled using Sequencher 4.7. Protein-coding genes, ribosomal and transfer RNA genes were identified by comparison with mitochondrial genomes of domestic bovids.
2.2 Phylogenetic analyses
Of the 339 mitochondrial genomes of Cetartiodactyla extracted from the EMBL/DDBJ/Genbank databases (January 2010), 77% were sequenced from domesticated animals, including Bos taurus (145 sequences), Sus scrofa (95), Ovis aries (8), Bos grunniens (4), Bubalus bubalis (3), Capra hircus (1), Camelus bactrianus (1), Camelus dromedarius (1), Lama glama (1), and Vicugna pacos (1). In order to reduce the size of our data matrix, only a few haplotypes were selected for domestic species. Our final dataset contains 210 sequences, representing 107 genera and 183 species of the order Cetartiodactyla. Three genera belonging to three different laurasiatherian orders were used as outgroups to root the cetartiodactyl tree: Pteropus (Chiroptera), Equus (Perissodactyla) and Panthera (Carnivora).
The 210 complete mitochondrial genomes were aligned by eye using Se-Al v2.0a11 [24]. All ambiguous regions, i.e., involving ambiguity for the position of the gaps, were excluded from the analyses to avoid erroneous hypotheses of primary homology. For this reason, the control region was not included in the alignment because of the presence of many ambiguous gaps. The final alignment consists of 14,902 nucleotide sites (nt) (Appendix 3).
The Maximum Likelihood (ML) analyses were done in RAxML-7.0.4 [25], with bootstrap percentages (BP) computed after 1000 replicates using the GTR + CAT model for the rapid bootstrapping algorithm.
The ML analysis of the total alignment of 14,902 nt was performed using 36 partitions corresponding to 12S (811 nt), 16S (1,274 nt), all non-coding regions and tRNAs (1461 nt), and the three codon-positions of the 11 following protein genes: ND1 (957 nt), ND2 (1044 nt), CO1 (1545 nt), CO2 (684 nt), ATP8 + ATP6 (841 nt), CO3 (786 nt), ND3 (357 nt), ND4L + ND4 (1670 nt), ND5 (1821 nt), ND6 (511 nt), and Cytb (1140 nt) (overlapping protein-coding genes were analyzed together, i.e., ATP8 + ATP6 and ND4L + ND4).
The ML analyses were also done on 10 half-overlapping sub-datasets (i–x) of the same length, i.e. 2980 nt, corresponding to the following positions: (i) 1–2980; (ii) 1491–4470; (iii) 2981–5960; (iv) 4471–7450; (v) 5961–8940; (vi) 7451–1,0430; (vii) 8941–11,920; (viii) 10,431–13,410; (ix) 11,921–14,900; (x) 13,411–14,902 + 1–1488. The use of half-overlapping sub-datasets implies that all nucleotide sites of the total mtDNA alignment are represented twice in these ML analyses. The lists of bipartitions obtained from BP analyses of the 10 sub-datasets of 2980 nt were transformed into a weighted binary matrix for supertree construction using the SuperTRI method [26]. Each binary character corresponds to a node, which was weighted according to its frequency of occurrence in one of the 10 lists of bipartitions. In that way, the SuperTRI method takes into account both principal and secondary signals, because all phylogenetic hypotheses found during the 10 BP analyses are represented in the weighted binary matrix used for supertree construction. The reliability of the nodes was then assessed using two measures: the supertree bootstrap percentages (SBP) were obtained from PAUP 4* version 4b10 [27] after 1000 BP replicates of the matrix of 8967 binary characters; and the mean bootstrap percentages (MBP) were directly calculated on SuperTRI. The SBP and MBP values were reported on the best ML tree found with the total alignment of 14,902 nt (Fig. 1). Here, the SuperTRI analyses were conducted to test for phylogenetic signal in the mtDNA genome. If a robust node of the ML tree (BP > 80%) is recovered with high SBP and MBP values (BP > 80%), we can conclude that the signal is present all along the mtDNA genome. If a node of the ML tree is recovered with low MBP values (< 50%), we can conclude that the signal is weak or confined to a few fragments of the mtDNA genome. If there is a robust topological conflict between ML and SuperTRI results, we can infer that at least one of the studied genomes has been partially contaminated by a DNA sequence of Numt or that of another species [28].
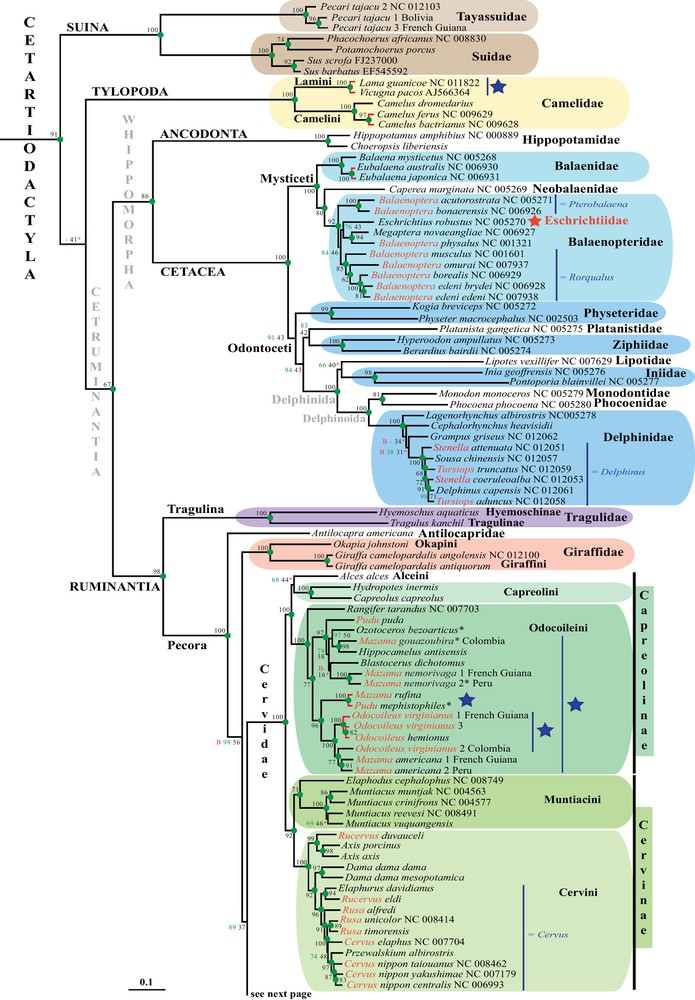
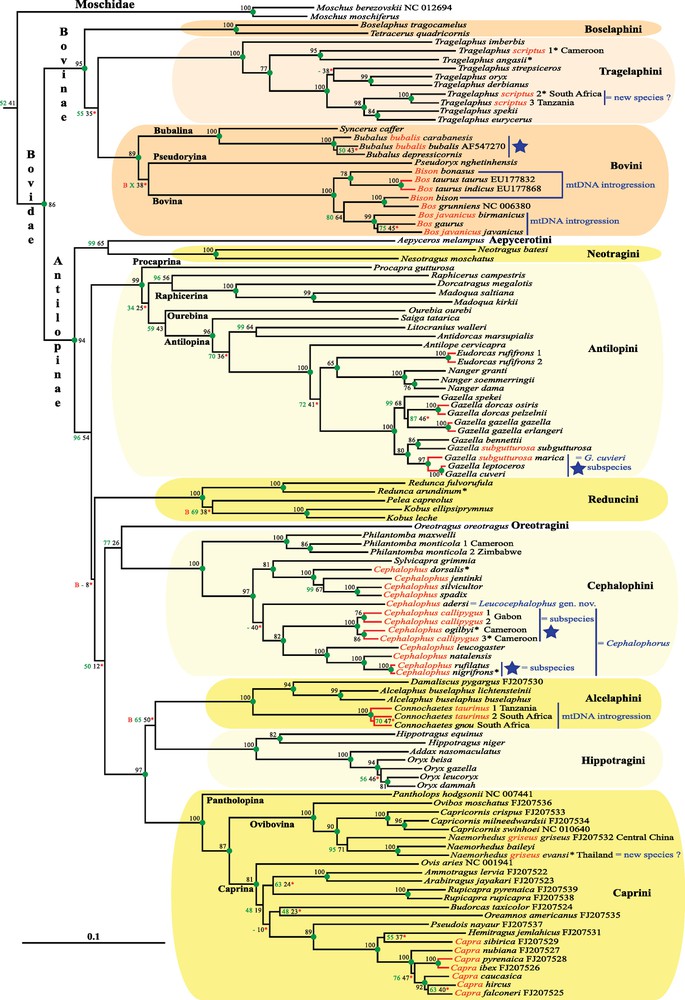
Phylogeny of Cetartiodactyla based on complete mitochondrial genomes. The tree is the ML phylogramm inferred with RAxML. Green values on the branches are bootstrap percentages (BP) obtained from the analysis of the alignment including 14,902 nucleotide unambiguous characters. Green circles indicate nodes that are supported by BP = 100 and recovered in the 50% Bayesian consensus tree (PP > 0.5). A red “B” indicates that the node was in conflict with the 50% Bayesian consensus tree. Two symbols were used for nodes that were not found in the BP majority-rule consensus trees: “X” indicates that another hypothesis is strongly supported in the BP analyses (i.e., BP > 80%); whereas “-” indicates that the alternative hypothesis is not robust (i.e., BP < 80%). Black values on the branches are Mean BP (MBP) calculated with the SuperTRI method and using the independent BP analyses of 10 overlapping sub-datasets of 2980 nucleotide sites (see Materials and methods). A red asterisk indicates that the node was not robust after 1000 BP replicates of the SuperTRI matrix (i.e., SBP < 80%). Red branches indicate the taxa that differ by a nucleotide distance of less than 2%. Non-monophyletic taxa are highlighted in red. Some taxonomic propositions are written in blue at the right of the taxa. Taxa labelled by a blue star are those that require additional molecular and morphological investigations to provide definitive conclusions. A species name labelled with an asterisk indicates that the sample was collected from local hunters, suggesting the possibility of species misidentification. Masquer
Phylogeny of Cetartiodactyla based on complete mitochondrial genomes. The tree is the ML phylogramm inferred with RAxML. Green values on the branches are bootstrap percentages (BP) obtained from the analysis of the alignment including 14,902 nucleotide unambiguous characters. Green circles ... Lire la suite
Bayesian phylogenetic reconstructions were conducted with PhyloBayes 3.1 [29] under the CAT-GTR + Gamma 4 site-heterogeneous mixture model applied to the concatenated nucleotide dataset. Two independent MCMC chains were run for a total of 15,000 cycles (equivalent to 4.5 million generations) sampling trees and parameters every ten cycles. The 50% Bayesian consensus tree was computed from the 2000 trees sampled by the two chains after removing the first 500 trees of each chain corresponding to the burnin stage. The maximum difference in clade posterior probabilities between the two chains was 0.08, which indicates that the two chains have independently converged towards similar posterior distribution of trees.
2.3 Molecular datings
Divergence dates were estimated using a Bayesian relaxed molecular clock approach implemented in PhyloBayes 3.1 [29] using the ML topology of Fig. 1. All absolute ages of the geological periods and chronostratigraphic references were taken from the 2009 Geological Time Scale of the Geological Society of America (available at http://www.geosociety.org/science/timescale/). For fossil calibrations, we selected six time constraints:
- (1) within Cetacea, the split between Mysticeti and Odontoceti, which is well-documented in the fossil record, was set at 35 ± 1 Ma (Million years ago) [4];
- (2) the first occurrence of the Bovidae, which is also well-documented in the Early Miocene of Africa and Eurasia was set at 20 ± 2 Ma [30];
- (3) the split between Cervini and Muntiacini was set to 9 ± 1 Ma, based on fossils from the Late Miocene of China, such as Cervavitus shanxius and Muntiacus noringenensis [31,32];
- (4) the age of extant Odocoileini was set at 5 ± 1 Ma, as the synapomorphy of the tribe, i.e., the presence of a vomerine septum dividing the choana, appears in Pavlodaria from the Early Pliocene [33];
- (5) the age for the divergence between cetaceans and hippos was set at 55 ± 5 Ma, as the oldest known cetaceans are from the Early Eocene of the Indo-Pakistan region (50–52.5 Ma) [4,5];
- (6) the common ancestor for Choeropsis and Hippopotamus was dated to 8 ± 1 Ma, as modern-looking hippopotamids appeared suddenly in the Late Miocene of Africa and Arabia at around 7.5 Ma [34]. The prior on the root of the tree, i.e., Scrotifera, was set at 80 Ma, in agreement with molecular studies of placental mammals [35,36].
As recommended by Lartillot et al. [29], the analyses were conducted using the CAT-GTR + Gamma 4 model and a log-normal autocorrelated clock relaxation model. However, we tested three different models, namely UNI-HARD, BD-HARD and BD-SOFT, which used different priors: uniform (UNI) versus birth–death (BD) priors on divergence times, combined with either hard or soft fossil calibrations [37]. We also performed molecular datings using five of the 6 fossil constraints in a leave-one-out approach aiming at estimating the robustness to calibration settings. For the excluded constraint, the molecular and fossil estimated divergence dates were compared. This was done for each of the six constraints. All calculations were conducted by running a MCMC chain for 30,000 cycles sampling posterior rates and dates every 10 cycles. Posterior estimates of divergence dates were then computed from the last 2500 samples accounting for the initial burnin period.
3 Results
3.1 Intra- and interspecific variations
Nucleotide distances (Appendix 4) were calculated on three alignments: the data matrix used for phylogenetic analyses, referred to as “mtDNA” (14,902 nt), and two protein-coding genes, i.e., CO1 (1542 nt), because this marker is currently used as reference DNA sequence in many DNA Barcoding projects; and Cytb (1140 nt), because many sequences of this gene are available in the nucleotide databases.
Similar nucleotide distances were found for mtDNA and CO1 sequences, while higher values were generally observed for Cytb. For most species, intra-specific comparisons show low levels of nucleotide variation, with mtDNA and CO1 distances < 3% and Cytb < 4% (e.g., Bos taurus, Cervus nippon, Gazella dorcas, G. gazella, and Giraffa camelopardalis).
Surprisingly, very low mtDNA sequence distances (< 2%) were detected for several interspecific comparisons: Mazama rufina versus Pudu mephistophiles; Camelus bactrianus versus C. ferus; Capra ibex versus C. pyrenaica; Cephalophus callipygus versus C. ogylbi; Cephalophus nigrifrons versus C. rufilatus; Connochaetes gnou versus C. taurinus; Eudorcas rufifrons versus E. thomsonii; Eubalaena australis versus E. japonica; between Gazella cuveri, G. leptoceros, and G. subgutturosa marica; Lama guanicoe versus Vicugna pacos; and Odocoileus hemionus versus O. virginianus.
In two cases, we detected a high percentage of nucleotide divergence between populations (> 5%), which may indicate the existence of two different species rather than subspecies. In Tragelaphus scriptus, the bushbuck from Tanzania and South Africa differ by 2.8% in their mtDNA, whereas they are highly divergent from the bushbuck of Cameroon (9.4–9.6%). A high sequence distance was also found between Chinese and Thai gorals (Naemorhedus griseus) (6.9%).
3.2 Mitogenomic phylogeny of Cetartiodactyla
The ML analyses of the alignment of 14,902 characters and 210 taxa produced the phylogenetic tree presented in Fig. 1. The topology of the Bayesian tree was very similar (Appendix 5). All the five suborders of Cetartiodactyla are recovered with maximum support values (BP = 100; PP = 1): Ancodonta, Cetacea, Ruminantia, Suina, and Tylopoda. They are also highly supported by the SuperTRI analyses of the 10 overlapping sub-datasets of 2980 nt (SBP = 100; MBP = 98–100), indicating that the phylogenetic signal is present all along the mtDNA genome. In addition, we found several molecular signatures (non-homoplastic synapomorphies), which can be used to characterize each of the five suborders (Appendix 5): Ancodonta (35), Cetacea (11), Ruminantia (3), Suina (8), and Tylopoda (21). The Whippomorpha clade, which includes Cetacea and Ancodonta (Hippopotamidae), is strongly supported by our analyses (BP/SBP = 100; MBP = 86; PP = 1). The Cetruminantia clade, which unites the three suborders Ancodonta, Cetacea, and Ruminantia, is also well supported by the analyses (BP/SBP = 100; MBP = 67; PP = 1). The inter-relationships between Cetruminantia, Suina, and Tylopoda are not stable. The ML and Bayesian topologies show a basal position for Suina (Fig. 1A), whereas bootstrap analysis favors an early divergence of Tylopoda (BP = 65 versus BP = 23). The SuperTRI analyses indicate that both topologies have similar support values (MBP/SBP = 41/57 and 39/43 for a basal position for Suina and Tylopoda, respectively).
Within Suina, both Suidae and Tayassuidae are found to be monophyletic (BP/SBP/MBP = 100; PP = 1). Within Suidae, the two genera endemic to sub-Saharan Africa, Phacochoerus and Potamochoerus, are found to be sister groups (BP = 100; SBP = 98; MBP = 74; PP = 1).
Within Tylopoda, the monophyly of Camelus is highly supported, as well as the sister–group relationship between Lama and Vicugna (BP/SBP/MBP = 100; PP = 1).
Within Cetacea, the basal division between Mysticeti and Odontoceti is recovered. The monophyly of Odontoceti is however less robust than that of Mysticeti (BP/SBP/MBP/PP = 91/96/43/0.93 versus 100/100/100/1). Within Mysticeti, the family Balaenidae is found to be monophyletic with maximum support, and its early divergence is also highly supported (BP/SBP = 100; MBP = 80; PP = 1). The family Balaenopteridae is paraphyletic, because of the inclusive position of Eschrichtius (family Eschrichtiidae). In addition, the genus Balaenoptera is paraphyletic, because B. acutorostrata and B. bonaerensis are the sister–group of a clade containing Eschrichtius, Megaptera, and other species of Balaenoptera (BP = 84; SBP = 96; MBP = 46; PP = 0.99). Within Odontoceti, all families are found to be monophyletic with maximum support values. Most inter-familial relationships are supported by BP and SBP > 80, but only three clades have MBP > 50: (i) Monodontidae + Phocoenidae (MBP = 81), (ii) their association with Delphinidae (MBP = 100); and (iii) Delphinida, i.e., the clade uniting Iniidae and Lipotidae with the three latter families (MBP = 100). The position of Lipotes (Yangtze River dolphin) as sister–group of the Amazon River dolphins (family Iniidae) is not robut (BP = 66; SBP = 53; MBP = 40; PP = 0.57). Within Delphinidae, the genera Stenella and Tursiops are found to be polyphyletic (BP/SBP = 100; MBP = 91–98; PP = 1). Within the group containing species of Delphinus, Sousa, Stenella and Tursiops, all terminal branches are very short, suggesting recent divergences. In agreement with that result, molecular dating estimates suggest that all events of speciation occurred during the Pleistocene epoch, between 1.81 ± 0.6 Ma and 0.84 ± 0.3 Ma (Fig. 2).
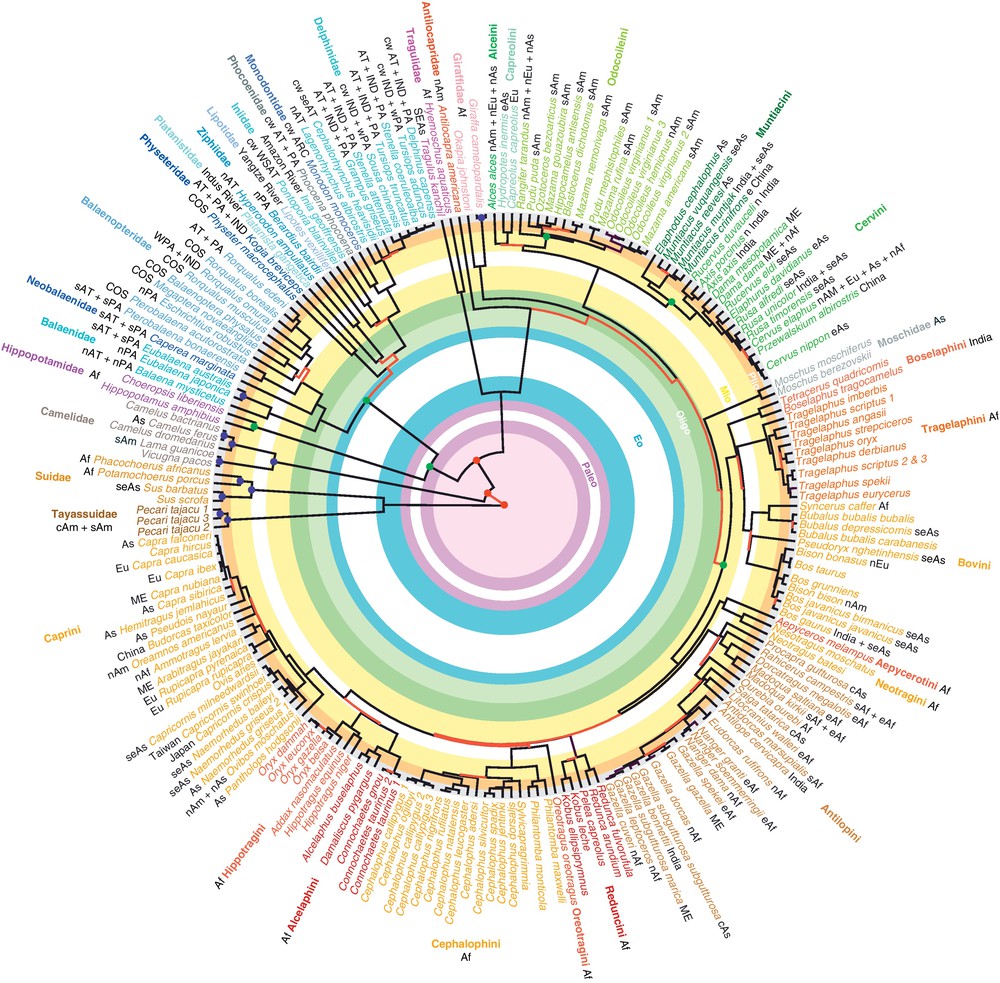
A molecular timescale for Cetartiodactyla. The estimates of divergence time were calculated under PhyloBayes 3.1 using the BD-SOFT model on the ML topology (see details in Materials and methods). Green discs indicate the six calibration points used for molecular estimation. Red discs indicate the nodes concerned by unreliable estimations of divergence time, i.e., for which no overlapping values were found with the two other Bayesian dating models (BD-HARD and UNI-HARD). Blue discs indicate the nodes with a high incertitude, i.e., for which the standard deviation exceeds 50% of the mean age. The red branches indicate the relationships that were found not to be robust in both mtDNA matrix and SuperTRI analyses (BP < 95 or PP < 0.75; MBP < 50) or that were conflicting with the 50% Bayesian consensus tree. The following abbreviations were used for indicating the geographic distribution: c (central), n (North), s (South), e (East), w (West); Eu (Europe), Af (Africa), Am (America), As (Asia), and ME (Middle East) for terrestrial cetartiodactyls; ARC (Arctic Ocean), AT (Atlantic Ocean), PA (Pacific Ocean), IND (Indian Ocean), ANT (Antarctic Ocean), COS (Cosmopolitan), and cw (coastal waters) for Cetacea. Masquer
A molecular timescale for Cetartiodactyla. The estimates of divergence time were calculated under PhyloBayes 3.1 using the BD-SOFT model on the ML topology (see details in Materials and methods). Green discs indicate the six calibration points used for molecular ... Lire la suite
Within Ruminantia, the families Bovidae, Cervidae, Giraffidae, Moschidae, and Tragulidae are highly supported by the analyses (BP/SBP = 100; MBP = 86–100; PP = 1). Most inter-familial relationships are supported by BP and SBP > 80, but only two clades have MBP > 50: (i) the infra-order Pecora, which groups all ruminant families except Tragulidae (MBP = 100; PP = 1); and (ii) the early offshoot of Antilocapridae within Pecora (MBP = 56). The clade uniting Bovidae, Cervidae and Mochidae is robust in the BP analysis of the mtDNA matrix (BP = 89) and that of the SuperTRI matrix (SBP = 88), but the low MBP value (37) reveals that the signal is only present on a few portions of the mtDNA genome. The Bayesian analysis of the mtDNA matrix rather supports a sister–group relationship between Antilocapridae and Giraffidae (PP = 0.99). The position of Moschidae is unstable: although the best hypothesis suggests a sister–group relationship with Bovidae (BP = 52; SBP = 82; MBP = 41; PP = 0.73), an alternative association with Cervidae cannot be completely excluded by the data (BP = 46; SBP = 18; MBP = 30). Within Bovidae and Cervidae, all supra-generic taxa described in previous studies [18,22,38,39] are highly supported (BP = 100; SBP = 99–100; MBP = 71–100; PP = 1), i.e., the subfamily Bovinae and the three tribes Bovini, Boselaphini, and Tragelaphini; the subfamily Antilopinae and the tribes Antilopini, Alcelaphini, Caprini, Cephalophini, Hippotragini, Neotragini, Reduncini; the subfamily Cervinae and the tribes Cervini and Muntiacini, and the subfamily Capreolinae and the tribes Capreolini and Odocoileini. Most intertribal relationships within Bovinae, Antilopinae and Capreolinae are not robust, suggesting rapid diversifications. However, we found three robust nodes within the Antilopinae: the sister–group relationship between Aepycerotini and Neotragini (BP = 99; SBP = 100; MBP = 65; PP = 1), their basal divergence from other tribes (BP = 96; SBP = 100; MBP = 54; PP = 1), and the clade composed of Alcelaphini, Caprini, and Hippotragini (BP/SBP = 100; MBP = 97; PP = 1).
Several new robust relationships between genera of Ruminantia were detected in this study. Within Odocoileini, South American deer falls into two divergent clades: the first one includes Blastocerus, Hippocamelus, Ozotoceros, Pudu puda, and two species of Mazama, M. gouazoubira and M. nemorivaga (BP/SBP = 100; MBP = 97; PP = 1); the second one groups Odocoileus, Pudu mephistophiles and two other species of Mazama, M. americana and M.rufina (BP/SBP = 100; MBP = 96; PP = 1). The monophyly of two genera of Odocoileini is therefore questioned by our data: Mazama and Pudu. In addition, the species Odocoileus virginianus is found to be paraphyletic because one specimen from Colombia appears as sister–group of M. americana (BP/SBP = 100; MBP = 77; PP = 1). Within Cervini, three genera are found to be poly- or paraphyletic: Cervus, as C. nippon is the sister–group of Przewalskium (BP/SBP = 100; MBP = 97; PP = 1) with C. elaphus at the outside; Rucervus, as R. duvauceli is related to Axis (BP/SBP = 100; MBP = 99; PP = 1), whereas R. eldi is allied to Elaphurus (BP/SBP = 100; MBP = 94; PP = 1); Rusa, as R. alfredi is the sister–group of a large clade uniting the two other Rusa species with all species of Cervus and Przewalskium (BP/SBP = 100; MBP = 91; PP = 1). Within Antilopini, Dorcatragus is associated to Madoqua (BP/SBP/MBP = 100; PP = 1), and they are linked to Raphicerus (BP = 96; SBP = 100; MBP = 56; PP = 1); Antidorcas and Litocranius are sister-genera (BP/SBP = 99; MBP = 64; PP = 1), and they are included in a large clade with Saiga and gazelles (BP/SBP = 100; MBP = 96; PP = 1). Within Cephalophini, Sylvicapra is robustly associated to the three “giant duiker” species (C. jentinki, C. spadix C. sylvicultor) and C. dorsalis (BP/SBP = 100; MBP = 81; PP = 1).
Eleven genera of Ruminantia are found to be para- or polyphyletic: Bos, Bison, Capra, Cephalophus, and Tragelaphus in the family Bovidae, as well as Cervus, Mazama, Odocoileus, Pudu, Rucervus, and Rusa in the family Cervidae. More surprising is the fact that eight species were found not to be monophyletic: one species of Cervidae, i.e., O. virginianus, and seven species of Bovidae, Bos javanicus, B. bubalis, C. callipygus, Connochaetes taurinus, Gazella subgutturosa, N. griseus and T. scriptus.
3.3 Molecular datings
We estimated a molecular timescale for cetartiodactyls based on the topology of Fig. 1 and using a Bayesian relaxed clock approach [29]. Three models were tested for estimating divergence times: UNI-HARD, BD-HARD, and BD-SOFT. The divergence times estimated with the three methods were very similar, except for the deepest nodes that are close to the root of the tree (Table 1). For instance, the estimate calculated for the origin of Cetartiodacyla was highly variable between the three models: 58.6 ± 3.2 Ma (UNI-HARD), 86.8 ± 11.5 Ma (BD-HARD), and 142.0 ± 71.3 Ma (BD-SOFT). Clearly, using a Birth-Death prior leads to unreasonably old date for these nodes. Therefore, we consider that all ages older than the Paleocene obtained using the BD prior were not reliable. In addition, all divergence dates estimated for inter-relationships within Camelidae, Suidae, and Tayassuidae were characterized by a high uncertainty, i.e. with a standard deviation > 50% of the mean age.
Molecular dating of the main splitting events within Cetartiodactyla.
| BD-SOFT Mean |
SD | BD-HARD Mean |
SD | UNI-HARD Mean |
SD | Geologic Period | |
| Cetartiodactyla | 86.8 | 11.5 | 142.0 | 71.3 | 58.6 | 3.2 | Late Cretaceous |
| First divergence of Suina | 80.3 | 8.8 | 102.2 | 37.6 | 57.3 | 3.1 | Late Cretaceous |
| Cetruminantia | 69.7 | 4.7 | 69.0 | 8.2 | 55.1 | 2.6 | K/T limit |
| Whippomorphaa | 60.1 | 1.7 | 57.7 | 2.3 | 52.1 | 2.0 | Paleocene |
| Ruminantia | 51.6 | 4.9 | 50.1 | 6.3 | 46.8 | 3.2 | Early Eocene |
| Suidae + Tayassuidae | 49.1 | 11.0 | 67.2 | 33.4 | 45.8 | 4.4 | Paleocene–Middle Eocene |
| Cetaceaa | 34.4 | 0.5 | 34.6 | 0.6 | 34.5 | 0.5 | Eocene/Oligocene limit |
| Odontocetii | 31.9 | 1.7 | 34.6 | 0.6 | 33.1 | 1.0 | Early Oligocene |
| Pecora | 27.6 | 3.8 | 28.1 | 4.5 | 29.9 | 3.3 | Early Oligocene–Late Oligocene |
| Mysticetii | 25.2 | 2.8 | 25.1 | 3.7 | 29.4 | 1.9 | Late Oligocene–Early Miocene |
| Pecora but Antilocapridae | 25.2 | 3.1 | 25.7 | 3.4 | 27.6 | 2.4 | Late Oligocene–Early Miocene |
| Bovidae + Moschidae + Cervidae | 23.4 | 2.6 | 23.8 | 2.8 | 25.6 | 1.6 | Late Oligocene–Early Miocene |
| Moschidae + Bovidae | 22.4 | 2.4 | 22.7 | 2.5 | 24.3 | 1.2 | Late Oligocene–Early Miocene |
| Balaenopteridae sensu lato + Neobalaenidae | 22.3 | 3.2 | 22.3 | 3.9 | 27.1 | 2.5 | Late Oligocene–Early Miocene |
| Physeteridae | 21.9 | 3.6 | 21.7 | 3.7 | 25.8 | 2.8 | Late Oligocene–Early Miocene |
| Tragulidae | 21.8 | 6.6 | 22.5 | 7.2 | 24.3 | 6.2 | Late Oligocene–Early Miocene |
| Delphinida | 20.5 | 3.2 | 20.0 | 3.7 | 24.2 | 2.7 | Early Miocene |
| Iniidae + Lipotidae | 19.9 | 3.2 | 19.4 | 3.8 | 23.7 | 2.9 | Early Miocene |
| Bovidaea | 19.7 | 1.9 | 20.0 | 1.9 | 21.5 | 0.5 | Early Miocene |
| Camelidae | 17.3 | 8.0 | 16.0 | 7.9 | 16.0 | 7.9 | Late Oligocene–Late Miocene |
| Antilopinae | 17.0 | 1.8 | 17.5 | 2.2 | 19.6 | 0.8 | Early Miocene–Middle Miocene |
| Ziphiidae | 16.6 | 3.5 | 16.4 | 3.8 | 20.2 | 3.6 | Early Miocene–Middle Miocene |
| Bovinae | 16.4 | 2.0 | 16.8 | 2.2 | 18.9 | 1.1 | Early Miocene–Middle Miocene |
| Antilopinae–(Aepycerotini + Neotragini) | 16.0 | 1.8 | 16.5 | 2.2 | 18.8 | 0.9 | Early Miocene–Middle Miocene |
| Balaenopteridae sensu lato | 15.5 | 3.0 | 15.8 | 3.6 | 20.6 | 3.4 | Early Miocene–Middle Miocene |
| Aepycerotini + Neotragini | 15.3 | 1.9 | 15.7 | 2.3 | 18.0 | 1.2 | Early Miocene–Middle Miocene |
| Giraffidae | 15.2 | 2.9 | 15.7 | 3.3 | 17.2 | 3.3 | Early Miocene–Middle Miocene |
| Cephalophini + Oreotragini | 14.4 | 1.7 | 15.0 | 2.4 | 17.6 | 1.1 | Middle Miocene |
| Inia + Pontoporia | 14.0 | 3.0 | 13.7 | 3.6 | 17.4 | 3.5 | Early Miocene–Middle Miocene |
| Bovini | 13.3 | 1.9 | 13.8 | 2.4 | 16.1 | 1.6 | Middle Miocene |
| Delphinidae + Monodontidae + Phocoenidae | 13.2 | 2.9 | 13.2 | 3.2 | 17.5 | 3.6 | Middle Miocene |
| Alcelaphini + Hippotragini + Caprini | 12.5 | 1.8 | 13.1 | 2.4 | 15.7 | 1.2 | Middle Miocene |
| Alcelaphini + Hippotragini | 12.0 | 1.9 | 12.5 | 2.4 | 15.3 | 1.3 | Middle Miocene |
| Antilopini | 12.02 | 1.6 | 12.5 | 2.2 | 15.1 | 1.3 | Middle Miocene |
| Suidae | 11.7 | 6.5 | 16.1 | 10.9 | 18.6 | 8.2 | Early Miocene–Late Miocene |
| Balaenidae | 11.5 | 3.9 | 12.0 | 4.2 | 17.3 | 4.8 | Middle Miocene–Late Miocene |
| Cervidae | 10.7 | 1.0 | 10.9 | 1.1 | 11.5 | 1.1 | Middle/Late Miocene limit |
| Monodontidae + Phocoenidae | 10.4 | 2.6 | 10.6 | 2.8 | 14.5 | 3.6 | Middle Miocene–Late Miocene |
| Odocoileinae | 9.2 | 0.8 | 9.3 | 0.9 | 10.0 | 1.1 | Late Miocene |
| Caprini | 9.2 | 1.8 | 9.8 | 2.3 | 12.5 | 1.4 | Late Miocene |
| Reduncini | 9.1 | 1.4 | 9.6 | 1.9 | 12.0 | 1.8 | Late Miocene |
| Alceini + Capreolini | 8.9 | 0.9 | 8.9 | 0.9 | 9.6 | 1.0 | Late Miocene |
| Cephalophini | 8.9 | 1.2 | 9.8 | 2.1 | 12.3 | 1.6 | Late Miocene |
| Cervinaea | 8.8 | 0.9 | 9.0 | 0.9 | 9.1 | 0.9 | Late Miocene |
| Neotragini | 8.8 | 1.5 | 9.4 | 2.1 | 11.1 | 2.0 | Late Miocene |
| Antilopina | 8.5 | 1.3 | 9.1 | 1.9 | 11.3 | 1.5 | Late Miocene |
| Bubalina | 8.2 | 1.7 | 8.7 | 2.2 | 10.9 | 2.0 | Late Miocene |
| Hippopotamidaea | 8.1 | 1.0 | 8.0 | 0.9 | 8.1 | 0.9 | Late Miocene |
| Muntiacini | 7.4 | 0.9 | 7.6 | 1.0 | 7.7 | 1.0 | Late Miocene |
| Tragelaphini | 6.8 | 1.7 | 7.2 | 2.0 | 9.1 | 2.1 | Late Miocene |
| Hippotragini | 6.8 | 1.6 | 7.3 | 2.2 | 9.8 | 1.8 | Late Miocene–Pliocene |
| Boselpahini | 6.6 | 1.8 | 6.9 | 2.1 | 8.8 | 2.4 | Late Miocene–Pliocene |
| Alcelaphini | 6.0 | 1.5 | 6.6 | 2.0 | 8.8 | 1.9 | Late Miocene–Pliocene |
| Cervini | 5.9 | 0.8 | 6.1 | 1.0 | 6.3 | 1.0 | Miocene/Pliocene limit |
| Odocoileinia | 5.7 | 0.4 | 5.6 | 0.4 | 5.7 | 0.3 | Miocene/Pliocene limit |
| Capreolini | 5.6 | 0.7 | 5.6 | 0.8 | 6.0 | 1.0 | Miocene/Pliocene limit |
| Tayassuidae | 5.2 | 3.6 | 7.5 | 6.1 | 12.3 | 9.1 | Miocene/Pliocene limit |
| Delphinidae | 5.1 | 1.4 | 5.3 | 1.6 | 8.9 | 3.4 | Miocene/Pliocene limit |
| Moschidae | 4.2 | 1.3 | 4.4 | 1.5 | 5.6 | 2.4 | Pliocene |
| Bovina | 3.3 | 0.9 | 3.7 | 1.3 | 5.0 | 1.8 | Pliocene |
a Calibration points.
Our alignment of complete mtDNA genomes provided 7449 parsimony informative characters for studying cetartiodactyl relationships. This large quantity of phylogenetic information explains why most deep and recent nodes of the tree (Fig. 1) were supported by maximum BP and PP values (75% of the nodes), as well as by high MBP values (68% of the nodes have MBP > 80%). However, all the nodes of the tree supported by BP < 95% were considered here as not being statistically robust, because they were systematically found to be poorly supported by the SuperTRI method (MBP < 50%). All these non-robust relationships (20% of the nodes) were highlighted in red in Fig. 2 in order to evidence concomitant events of rapid diversification. The hypothesis of rapid radiation can be satisfied only if the nodes both after and before the event are robustly supported. Using this criterion, we detected four main phases of radiation in the order Cetartiodactyla: the sudden occurrence of the three extant lineages within Cetartiodactyla (Cetruminantia, Suina and Tylopoda); the basal diversification of Cetacea during the Early Oligocene; and two radiations that concern both Pecora and Cetacea, one at the Oligocene/Miocene boundary and the other one in the Middle Miocene. In addition, we show that the high species diversity now observed in the families Bovidae and Cervidae built up during the Late Miocene and Plio-Pleistocene.
4 Discussion
4.1 Alpha-taxonomic issues
The mtDNA distances between several species studied here are lower than those generally obtained between subspecies (< 2%; red branches in Fig. 1). Four main hypotheses can be proposed for explaining the existence of similar mtDNA genomes in two putative species. The first two hypotheses propose that the samples were extracted from individuals of the same species, because of either species misidentification (human error) or imperfect taxonomy (species synonymy). The third hypothesis implies that the mtDNA genome of one species was transferred into the other by inter-specific hybridization, a process known as mtDNA introgression. The fourth hypothesis suggests a recent speciation event.
The possibility of species misidentification cannot be completely ruled out for bushmeat or skin samples collected by hunters. In such cases, the risk increases if two or several cryptic species live in the same geographic area. In our dataset, we are aware that a few samples taken from African forest duikers (Cephalophini) and South American dwarf deer (Odocoileini) may have been misidentified (indicated by an asterisk in Fig. 1).
A first example concerns Mazama rufina and Pudu mephistophiles, which share very similar mtDNA genomes (98.6%). The hypothesis of species assignation error for one of the two samples cannot be totally dismissed because our sequences were obtained from pieces of skin collected in Colombia, a country where the same name “venado chonta” can be used to designate both species [40]. In addition, samples from the Colombian dwarf brocket (M. rufina) and northern pudu (P. mephistophiles) can be easily mistaken because both species live in the same habitat, i.e., the mountainous forests between 2000 and 4000 m in Colombia, Ecuador and Peru, and because they exhibit the same color pattern, with a reddish body that shifts to black on the face and lower part of the legs. However, Pudu mephistophiles is most readily distinguished from Mazama rufina by its smaller body size (weight: 5–5.8 kg versus 10–15 kg; shoulder height: 25–40 cm versus 45 cm) [40–42].
A second example of possible species assignation error concerns a bushmeat sample from Ogilby's duiker (Cephalophus ogilbyi), because its mtDNA sequence is closely related to that of a Cameroonian Peter's duiker (C. callipygus) (98.5%), which makes the species C. callipygus paraphyletic (Fig. 1B). The quarry was killed in the Korup National Park, in western Cameroon close to the Nigerian border, a geographic area where C. callipygus is expected to be absent [1]. Our analyses of nucleotide databases (Genbank and BOLD) revealed that the three species C. ogilbyi, C. callipygus and C. weynsi share similar mitochondrial sequences (12S rRNA: 98–99%; CO1: 97–98%; cytochrome b: 96–98%), suggesting subspecies rather than species rank status. In agreement with this proposition, the Weyn's duiker (C. weynsi) was treated as a subspecies of C. callipygus in several classifications [43,44], and Peter and Ogilby's duikers were considered to belong to the same species [45]. Similar nucleotide distances were also observed among four parapatric species of red duikers, C. nigrifrons, C. rufilatus, C. harveyi, and C. natalensis (12S rRNA: 98–99%; CO1: 97–99%; cytochrome b: 96–98%). Once again, C. harveyi was relegated to a subspecies of C. natalensis in several classifications [2,44]. All these results suggest that the alpha taxonomy and geographic ranges of red duiker species need to be reassessed using voucher specimens, and that nuclear DNA barcodes or microsatellites should be developed in order to evidence potential cases of mtDNA introgression (see below).
Several studies have shown that mitochondrial introgression can result in erroneous interpretation of the mtDNA topology for shallow phylogenies [46–49]. Indeed, natural hybridization between closely related species can lead to the transfer of mtDNA from one species into another, a process known as mtDNA introgression. By selection, the invaded mtDNA genome from the donor can then be fixed into the host species. Inter-specific hybridization can generally occur between closely related taxa for which prezygotic isolation (behavioural and mechanical) and postzygotic isolation (zygote mortality and hybrid inviability and sterility) have not been fully accomplished. Such mtDNA replacements have been inferred for the ancestors of goat species [48], European bison [50] and Cambodian banteng [49]. A similar case of mtDNA introgression can be also inferred to explain the paraphyly of C. taurinus with respect to C. gnou (Fig. 1B). The hypothesis is supported by the existence of a geographic clade containing South African specimens from two different species (C. gnou and C. taurinus). Actually, hybrids between C. gnou and C. taurinus are known to be fertile [51], and some hybrid animals have been identified on the basis of cranial morphology [52] and microsatellite analyses [53]. Hybridization may have occurred during the Pleistocene or Holocene epochs in the eastern regions of South Africa, where the two species are still found in sympatry. These animals are also extensively farmed and often held together on the same reserve. The hybridization could have thus also been due to recently human mediated activities. Similarly, mtDNA introgression can be also advanced to explain the paraphyly of O. virginianus (specimens 1 and 3) with respect to O. hemionus (Fig. 1A), as several cases of natural hybridization have been previously described in some populations of North American deer [54].
All these identified cases of mtDNA introgression suggest that the phenomenon is more frequent in gregarious than solitary species. One explanation is that the first step consists in the adoption of at least one young foreign female into a herd of another species [49]. Accordingly, we can assume lower levels of mtDNA introgression for a few groups of Cetartiodactyla in which all species show a solitary behavior, such as Tragulidae (chevrotains), Muntiacini (muntjacs), a few genera of Odocoileini (e.g., Mazama and Pudu), Cephalophini (duikers), Madoqua (dik-diks) and Raphicerus (grysboks and steenbok).
The Arabian sand gazelle was first described as a species, Gazella marica [55], and later treated as a subspecies of G. leptoceros [56]. More recently, the Arabian sand gazelle has been considered as a subspecies of G. subgutturosa (G. s. marica) based on morphological similarity [57]. However, our analyses show that G. subgutturosa is actually polyphyletic: G. s. marica is closely related to the North African species G. cuvieri and G. leptoceros, as recently found in the analysis of cytochrome b sequences [58], whereas the Asian subspecies G. s. subgutturosa is allied to G. bennettii. Actually, the morphology of the Arabian sand gazelle differs from that of Asian goitered gazelles: it has a white face that lacks markings; and females have well-developed horns (as in G. cuvieri and G. leptoceros), whereas females of the three other subspecies of G. subgutturosa (subgutturosa, hilleriana and yarkandensis) are hornless or have short horns [59]. As a consequence, both genetic and morphological data indicate that the Arabian sand gazelle should be removed from G. subgutturosa. In addition, the pairwise distances between the Arabian sand gazelle and North African gazelles are very low (< 1.5%), suggesting that the three species should be considered as subspecies of the same species G. cuvieri.
Several previous genetic studies have concluded that Swamp and River buffalo have distinct mtDNA and Y-chromosome haplotypes, suggesting that they belong to different subspecies (B. b. bubalis and B. b. carabanesis), which were domesticated independently [60,61]. Our analyses confirm that the mtDNA genomes of Swamp and River buffaloes are different (2.1%). However, a similar distance was also found with the anoa (2.3%), Bubalus depressicornis, an endemic species to Indonesia that is only found on Sulawesi and Buton islands [1]. These data therefore imply that the three taxa belong to three distinct species that recently diversified during the Pleistocene (Fig. 2; 1.3 ± 0.5 Ma): B. bubalis, B. carabanesis and B. depressicornis. This taxonomic proposition is also supported by cytogenetic differences between anoa (2n = 48, NFa = 58), Swamp buffalo (2n = 48, NFa = 56) and River buffalo (2n = 50, NFa = 58) [62,63].
Within T. scriptus, our mtDNA analyses show the existence of two very divergent lineages of bushbuck that differ by more than 9.4%: the first one is related to Tragelaphus angasii, and the second is the sister–group of T. eurycerus and T. spekii (Fig. 1B). Our result confirm the cytochrome b analyses of Moodley et al. [64], which have shown the existence of two geographic groups that may correspond to two distinct species, T. scriptus, which inhabits the north-western half of the sub-Saharan Africa, and T. sylvaticus, which is found in the south-eastern part of Africa. These taxonomic suggestions need to be confirmed by morphological and nuclear data.
A high distance was also found between the two subspecies of N. griseus (6.9%), suggesting the existence of a new cryptic species of goral in Thailand. The comparisons with all mtDNA sequences available for gorals in Genbank confirmed that the closest relative of the Thai goral is the Red goral (N. baileyi), as in Fig. 1B, a species found at higher elevations, at altitudes between 2000–4500 m, in northern Myanmar, China (southeast Tibet and Yunnan), and northeast India [1]. The mtDNA genome of the Thai goral differs by 4% from that of the Red goral, suggesting that the subspecies N. g. evansi could be elevated to a full species status (N. evansi). As the geographic distribution of N. baileyi and that of the two subspecies N. g. griseus and N. g. evansi appear to be uncertain [1], additional morphological and molecular data are needed to provide definitive conclusions on the taxonomic status of gorals found in north-western Thailand, but also in Myanmar, northeastern India, South China and extreme northern Vietnam.
The most spectacular taxonomic issue revealed by our data probably concerns deer of the tribes Odocoileini, where three genera are found to be non-monophyletic: Mazama, Odocoileus and Pudu (Fig. 1A). As previously mentioned, the South American deer needs major taxonomic revisions at the genus and species levels [39,65]. One of the most intriguing results is the paraphyly of Odocoileus, because of the sister–group relationship between Mazama americana and one of the three specimens of O. virginianus (Fig. 1A). The MRGOv14 sample was collected in 2001 by MRG on a male hunted in Vereda San Francisco (Nevado del Huila National Park, Colombia). Its mtDNA genome is divergent from all other South American deer (> 4.6% with Mazama americana, > 5.1% with other Odocoileus specimens), suggesting the possible existence of a new cryptic species in Colombia.
4.2 Towards a new beta taxonomy of the order Cetartiodactyla
The mitochondrial genome provides robust support for the monophyly of cetartiodactyl suborders, infra-orders, families, subfamilies and tribes described in previous classifications [1,10,18,39,66,67]. There is only one exception: the family Balaenopteridae, which is paraphyletic due to the inclusive placement of Eschrichtius robustus (family Eschrichtiidae) (Fig. 1A). This result was already found in the cetacean phylogeny of McGowen et al. [10] based on both mtDNA and nuclear genes. According to that, we consider that gray whales (E. robustus) should be included into the family Balaenopteridae.
Since the genus Balaenoptera is found to be paraphyletic, we propose to restrict the use of Balaenoptera to only one species, B. physalus, whereas other species should be arranged into Pterobalaena Eschricht, 1849 (type species Balaenoptera acutorostrata Lacépède, 1804) or Rorqualus F. Cuvier, 1836 (type species Balaena musculus Linnaeus, 1758) (Fig. 1A). Each genus is diagnosed by one unique substitution in the mtDNA alignment (Appendix 6): Pterobalaena by a transition T→C at pos. 7236 (tRNA-Lys), and Rorqualus by a transition G→A at pos. 1475 (rRNA-16S). Our study suggests that the four lineages corresponding to Balaenoptera + Megaptera, Eschrichtius, Pterobalaena and Rorqualus, diverged rapidly during the Middle Miocene, between 15.5 ± 3.0 Ma and 13.0 ± 2.7 Ma (Fig. 2). Surprisingly, hybridization between the blue whale (R. musculus) and the fin whale (B. physalus) is known to occur at least occasionally in both the North Atlantic and North Pacific Oceans [68]. The production of viable hybrids is probably exceptional for such divergent mammals. Indeed, interspecific hybridization usually occurs between closely related species that have diverged from each other quite recently, i.e., during the Pleistocene, and for which reproductive isolation has not already been fully achieved, e.g., between kouprey and banteng in Cambodia [49], between wolves and coyotes in North America [69], and between forest and savannah elephants in Africa [70]. In general, distant species cannot hybridize successfully because of pre-zygotic isolating barriers (such as the inability to copulate, and gamete transfer barriers) or post-zygotic barriers (hybrid inviability or hybrid sterility) [71]. However, mysticetes have nucleotide substitution rate that is approximately 10-fold slower than that of other mammals for both mitochondrial and nuclear genomes [72,73]. In addition, they have very similar karyotypes, with either 2n = 42 or 44 chromosomes [74]. The high conservation of genomic structure and sequences in mysticetes can therefore explain the occurrence of hybrids between blue and fin whales. However, the capacity of hybrids to produce viable offspring awaits confirmation [68]. To test possible mtDNA introgression events during the evolutionary history of mysticetes, we re-analyzed the alignment published in McGowen et al. [10] by excluding mtDNA markers (22 taxa and 28,800 characters). The phylogenetic tree obtained from nuclear genes (between 23 and 29 genes for each of the five balaenopterid genera; Appendix 7) confirms the monophyly of Pterobalaena (BP = 100), the sister–group relationship between Balaenoptera and Megaptera (BP = 100), and that between R. edeni and R. borealis (BP = 100), but all other relationships were found not to be robust (BP < 50) because of low levels of variation and a large amount of missing data. As these nuclear results are congruent with our mitochondrial tree and the Y-chromosome tree of Nishida et al. [19], we can safely conclude to the occurrence of restricted gene flow between B. physalus and R. musculus, suggesting that hybrids are sterile or have lower fitness.
Within the family Delphinidae, our mtDNA analyses show a polyphyletic pattern for both the genera Stenella and Tursiops (Fig. 1A). This result corroborates the signal obtained from nuclear markers [75,76]. The two genera belong to a species complex, which contains other genera, such as Delphinus and Sousa, and which has rapidly flourished during the Pleistocene (Fig. 2; 1.8 ± 0.6 Ma). All these points support the hypothesis of a recent radiation within the family Delphinidae. Therefore, from the taxonomic point of view, we agree with the proposal of LeDuc et al. [66] to synonymize Lagenodelphis, Stenella, Sousa and Tursiops with Delphinus.
Within Cephalophini, the genus Cephalophus is found to be paraphyletic with respect to Sylvicapra (Fig. 1B). This result contrasts with previous studies based on the mitochondrial cytochrome b gene, which found Sylvicapra as the sister–group of Cephalophus, with Philantomba being more divergent [77]. However, these relationships were not statistically supported (BP = 58). Here, the placement of Sylvicapra as sister–group of the giant duiker group (C. dorsalis, C. jentinki, C. silvicultor and C. spadix) is well supported (BP/SBP = 100; MBP = 81; PP = 1). If confirmed by nuclear data, this result may have direct taxonomic consequences, as it suggests that the genus Cephalophus should be split into four distinct genera: Cephalophus, now restricted to only giant duikers; the recognition of C. adersi as a distinct new genus, Leucocephalophus; the recognition of C. zebra as a distinct subgenus Cephalophula; and the placement of C. ogilbyi within the red duiker subgenus Cephalophorus. Alternatively, the genus Sylvicapra should be synonymised with Cephalophus.
Many taxonomic issues concern the genera of the family Cervidae (Fig. 1A): Cervus, Rusa, and Rucervus in the tribe Cervini, and Odocoileus, Mazama, and Pudu in the tribe Odocoileini. As proposed by Gilbert et al. [39], several species should be classified into the genus Cervus: Elaphurus davidianus, Przewalskium albirostris, Rucervus eldi, and all species of Rusa. As pointed out in previous studies [39,65], the taxonomy of South American deer is more problematic, and additional morphological and molecular data (including nuclear markers) are needed to understand the complex evolutionary history of the tribe Odocoileini.
4.3 No reliable divergence date estimates for basal relationships within Cetartiodactyla
Our phylogenetic analyses have produced conflicting signals for basal relationships within the Cetartiodactyla, suggesting that the diversification into three main lineages (Cetruminantia, Suina, and Tylopoda) occurred over a short period of time. Alternatively, this lack of stability at the base of the cetartiodactyl tree may be the consequence of three synergetic effects: the high levels of homoplasy due to the rapid evolution of the mitochondrial genome [78], the heterogeneity in evolutionary rates among taxa [72], and the use of divergent outgroups representing the orders Carnivora (Panthera), Chiroptera (Pteropus) and Perissodactyla (Equus).
The divergence dates estimated for the deepest nodes of the cetartiodactyl tree were found to be highly variable between the three relaxed molecular clock models (Fig. 2 and Table 1). In addition, these estimates became considerably older when the calibration point corresponding to the separation between hippos and cetaceans (55 ± 5 Ma) was removed. This problem may be due to the slower evolution of the mitochondrial genome of hippos and large whales (Appendix 8). In contrast, our molecular estimates of more recent divergences within Cetruminantia, i.e., which happened during the Cenozoic era, were found to be largely overlapping and compatible across the three relaxed molecular clock models, suggesting a positive effect resulting from the incorporation of several calibration points in this part of the tree, denser taxon sampling, and a large amount of unsaturated phylogenetic information.
4.4 Aquatic diversification of Cetacea
All the oldest cetacean fossils were extracted from the Early Eocene of the Indo-Pakistan region. They were terrestrial animals that foraged for food in freshwater. The earliest fully aquatic cetaceans originated at around 40 Ma with basilosaurids [4,5]. The basal diversification of crown cetaceans into mysticetes and odontocetes occurred at the “grande coupure” boundary, i.e., at the limit between Eocene and Oligocene (34.4 ± 0.5 Ma) [4]. Thereafter, our chronogram indicates that Odontoceti rapidly diversified into four lineages corresponding to Physeteridae, Platanistidae, Ziphiidae, and Delphinida during the Early Oligocene, between 31.9 ± 1.7 Ma and 28.6 ± 2.3 Ma. These molecular estimates fit well with the fossil record, which shows the sudden appearance of several lineages of toothed mysticetes and odontocetes during the Early Oligocene. This explosive radiation coincides with the transition from a greenhouse- to an icehouse-world at the Eocene/Oligocene boundary, and the tectonic opening of the Drake Passage between South America and Antarctica, which resulted in the formation of the Antarctic Circumpolar Current [79,80].
The toothless mysticetes, with baleen for filter feeding, are presumed to first occur in the Late Oligocene [5]. In agreement with these paleontological data, our analyses show that the three extant families of Mysticeti diverged from each other at the limit between Oligocene and Miocene, between 25.2 ± 2.8 Ma and 22.3 ± 3.2 Ma. Baleen whales primarily eat zooplankton, such as krill, copepods, and amphipods, as well as small fish. Interestingly, most extant krill genera first appeared before the end of the Oligocene [81], suggesting that their diversification just predated that of mysticetes. It seems however that other factors have influenced the early evolution of Mysticeti, because Odontoceti also diversified at roughly the same time, with the basal divergence between Physeter and Kogia within Physeteridae (21.9 ± 3.6 Ma), and the division of Delphinida into three lineages (between 20.5 ± 3.2 Ma and 19.9 ± 3.2 Ma) corresponding to Iniidae, Lipotidae, and the clade uniting Delphinidae, Monodontidae and Phocoenidae (superfamily Delphinoidea). The stratigraphic and phylogenetic analyses of Physeteridae have suggested that Physeter and Kogia are two surviving lineages of a wide radiation of sperm whales that took place during the Early Miocene [82,83]. The diversification of Mysticeti and Odontoceti at the Oligocene/Miocene boundary may have been triggered off by two global changes during this period: a warmer deep-sea water mass in both Atlantic and Pacific Ocean basins [84] and an important sea-level fall [85]. However, the fact that all families of Delphinida seem to appear later, i.e. in the late Middle Miocene to early Late Miocene [86], suggests, however, either a gap in the fossil record or an overestimated date for the basal diversification of Delphinida. Actually, although accommodated in principle by the relaxed-clock model, the higher rates of substitutions found in river dolphins (Appendix 8) may result into an inflated age estimate in the common ancestor of Iniidae, Lipotidae and Delphinoidea. Accelerated evolutionary rates in river dolphins may be explained by more frequent and severe bottlenecks in freshwater dolphin populations than in marine populations. Higher rates of substitutions are expected for species with evolutionary history punctuated by repeated bottleneck events because more substitutions go to fixation by drift in smaller populations [87]. This hypothesis should be further explored by studying the population genetics of river versus marine dolphin species of the family Delphinidae, such as S. fluviatilis versus S. guianensis and Orcaella brevirostris versus Orcaella heinsohni.
During the Middle Miocene, a second wave of diversification is documented in the fossil record of Cetacea [5]. In agreement with this observation, our molecular estimates suggest that this period was marked by many events of divergence at the base of Balenopteridae sensu lato (between 15.5 ± 3.0 Ma and 13.0 ± 2.7 Ma), Balenidae (11.5 ± 3.9 Ma), Ziphiidae (16.6 ± 3.5 Ma), between Inia and Pontoporia in South America (14.0 ± 3.0 Ma), and between Delphinidae and the common ancestor of Monodontidae and Phocoenidae (13.2 ± 2.9 Ma). After a long warming phase, from the Late Oligocene, around 26 Ma, until the Middle Miocene climatic optimum, between 17 and 15 Ma, a gradual cooling is well-documented from various oceanographic and terrestrial records [80,88]. Hamilton et al. [89] proposed that the ancestors of the four extant river dolphin genera independently colonized the shallow epicontinental seas that inundated the Amazon (Inia), Paraná (Pontoporia), Yangtze (Lipotes) and Indo-Gangetic (Platanista) river basins during the globally high sea levels of the Middle Miocene. Our molecular estimates are compatible with this scenario as the South American divergence between Inia and Pontoporia is dated at 14.0 ± 3.0 Ma.
Delphinids are now the most diverse and widespread family of the suborder Cetacea. According to our estimations, there was a rapid radiation of Delphinidae around the Miocene/Pliocene boundary (between 5.1 ± 1.4 Ma and 4.4 ± 1.2 Ma). Interestingly, the fossil record indicates that modern dolphins are absent in the Mediterranean and in the North Atlantic before the Early Pliocene [90]. Bianucci [91] proposed that the family Delphinidae had undergone an explosive radiation in the Pliocene of the Mediterranean, probably related to the recolonization of this basin after the Messinian salinity crisis. Our results suggest that the Early Pliocene radiation of delphinids was not confined to the Mediterranean, but was rather extended to all oceans and seas of the World. During the early Pliocene, high latitudes were warmer, continental ice sheets were largely absent from the northern Hemisphere, and the sea level was ∼ 25 m higher than today [92]. In addition, there was an enhanced hurricane activity [93]. All these climatic conditions may have favored the rapid diversification of Delphinidae. The genus Delphinus sensu lato (including Delphinus, Stenella, Sousa, Tursiops) has also intensively diversified during the Pleistocene. The fluctuations in sea level and water temperatures that characterized the Pleistocene epoch may have played a significant role in driving the speciation of extant dolphins.
4.5 Basal diversification of Ruminantia and Suina during the Early Eocene climatic optimum
Our molecular timescale suggests that the suborder Ruminantia split into Pecora and Tragulina during the early Eocene (51.6 ± 4.9 Ma). The diversification of Suina into Suidae and Tayassuidae may have occurred at more or less the same time (49.1 ± 11.0 Ma). Only a few fossils related to both Suidae and Tayassuidae were discovered in the Middle and Late Eocene of Central Asia and Southeast Asia [94,95]. However, Liping [96] suggests that extant Suina emerged in South Asia in the Middle Eocene or even earlier. By contrast, a large diversity of Ruminantia has been described from the Middle Eocene of Central Asia, Southeast Asia and North America [97,98]. Early ruminants were small forest dwellers, and probably mixed feeders with a diet similar to that of extant chevrotains, i.e., browsing herbivores eating mostly leaves, fruits and shoots. The first appearance of ruminants might be linked to the Early Eocene Climatic Optimum (EECO), between 53 and 50 Ma [98], which is consistent with our molecular datings. The climate changed dramatically during the EECO interval, with higher concentrations of greenhouse gases and a much warmer mean global temperature [99]. These changes promoted a major increase in floral and faunal diversity [100]. The palynological record indicates that rainforests extended into the mid-latitudes and that the two poles were forested [101,102]. Assuming an Asian origin of Ruminantia, as proposed by Métais et al. [97], these climatic conditions may have facilitated the rapid dispersal of early forest ruminants from Asia into North America through Beringia. By contrast, marine barriers, such as the Turgai Strait, which separated eastern Asia from Europe, and the North Sea [103], may have prevented an early Eocene migration of ruminants into Europe.
4.6 Rapid adaptive radiation of Ruminantia during the Late Oligocene and Early Miocene
The families of Pecora–Antilocapridae, Cervidae, Bovidae, Giraffidae, and Moschidae–diverged rapidly from each other at the Oligocene/Miocene boundary, between 27.6 ± 3.8 Ma and 22.4 ± 2.4 Ma. These estimates agree with the fossil record, which shows that most extant families of Pecora made their first appearance before 20 Ma [98]. During the Early Miocene, most habitats in western Europe and Africa were forested, whereas those of eastern Europe, Asia and North America were more open [98]. The spread of open-habitat grasses at the Oligocene/Miocene was promoted by a global environmental change, when the warming Late Oligocene was interrupted by a brief but deep glacial maximum at the Oligocene/Miocene boundary [104]. All these paleoecological data suggest that the emergence of modern pecoran families took place in the northern hemisphere, most probably in the eastern Eurasia. Then, the families dispersed and diversified rapidly: Antilocapridae and blastomerycine Moschidae entered in North America; Cervidae and other Moschidae stayed in Eurasia; Bovidae and Giraffidae appeared suddenly throughout the Old World [98].
According to the fossil record, two geographic lineages of the family Tragulidae also underwent a strong radiation during the Early Miocene: one in Africa and Europe, and another one in South Asia [105,106]. The discovery of tragulid remains in the Late Eocene of Thailand suggests that the family emerged and diversified firstly in Southeast Asia [97,107], and dispersed secondarily in other continents of the Old World [98]. Africa was isolated from the Mid-Cretaceous until the Early Miocene, when occasional connections were established through the Middle East with Eurasia [108]. The ancestor of the African branch of the family Tragulidae (today only represented by Hyemoschus) probably entered in Africa during the Early Miocene, as our molecular estimates indicate that African and Asian branches of the family Tragulidae split off at 21.8 ± 6.6 Ma. In favor of this scenario, the earliest tragulid fossils of Africa were dated between 20 and 22.5 Ma [105,109]. The fact that all extant chevrotains live in wet forested habitats suggests that rainforests occurred in East Africa, Europe and South Asia during the Early Miocene, as supported by palaeoecological studies [110–112].
4.7 Radiation of bovids during the Middle Miocene
The three tribes of Bovinae, i.e. Bovini, Boselaphini and Tragelaphini, diverged between 16.4 ± 2.0 Ma and 15.8 ± 2.0, whereas most tribes of Antilopinae emerged at the same time, between 16.0 ± 1.8 Ma and 14.5 ± 1.7 Ma, i.e. Antilopini, Cephalophini, Oreotragini, Reduncini, as well as Aepycerotini and Neotragini. At the current state of knowledge, the palaeontological data indicate that most tribes within the Bovidae appeared later, i.e., during the Late Miocene, between 7 and 10 Ma. The only exceptions are Antilopini and Reduncini, for which possible representatives have been described in the Middle Miocene of East Africa [113]. Our molecular estimates reveal therefore that a hiatus of 6–9 Myr exists in the fossil record of most bovid tribes.
The tribal radiation of Bovidae coincides with the Middle Miocene Climatic Optimum (MMCO), between 17 and 15 Ma, which was the last of a series of global warming events that have punctuated the Cenozoic [99]. This interval was the warmest time since 35 Ma in Earth's history. The warmer and more humid conditions prevailing during the Middle Miocene favored the expansion of evergreen forests in South and Central Europe [114], North Africa [110], north-western India [115], and in Asia, where they extended north to Korea and Japan [116]. The MMCO was directly followed by a drastic global cooling, from 15 to 13 Ma, marking a transition from a greenhouse world to an “icehouse-world” [99,117].
Previous biogeographical studies have supported an African origin for the last common ancestor of Antilopinae [22,118]. In Africa, the Middle Miocene climatic transition led to significant vegetation changes, with the expansion of open habitats and increased aridification in southwestern and eastern Africa [110]. These rapid changes created a mosaic of ecosystems in Africa, which may have promoted the emergence of antilopine tribes with different ecological preferences, such as Antilopini in open habitats, or Cephalophini and Neotragini in forested habitats. Interestingly, our molecular analyses indicate that the two extant species of Giraffidae, G. camelopardalis and Okapia johnstoni, diverged from each other during the Middle Miocene, at 15.2 ± 2.9 Ma. Today, the giraffe is associated with Acacia, Commiphora and Combretum savanna, whereas the okapi is endemic to the high canopy forests of DR Congo [1]. As in the case of Antilopinae, their common ancestor probably entered in Africa during the Early Miocene, and they adapted to different habitats during the Middle Miocene, the period when the two extant tribes–Giraffini and Okapini–diverged.
The diversification of Bovinae is more enigmatic, but most previous studies have proposed an early evolution in Eurasia [18,118,119]. Just two or three million years after the tribal radiation of Bovinae, the tribe Bovini rapidly diversified into the three subtribes Bovina, Bubalina and Pseudoryina during the late Middle Miocene, between 13.3 and 13.0 ± 2.0 Ma. The biogeographical analyses of Hassanin and Ropiquet [118] have supported an Asian origin for the tribe Bovini. Since all the three subtribes are currently more diversified in Southeast Asia than in other regions of the World, it seems more likely to consider that their common ancestor occurred in this area. A rapid diversification of Bovini during the late Middle Miocene of Southeast Asia is also corroborated by recent palaeoecological data, which document oscillations between tropical woodlands and grasslands in northern Thailand between 13.3 and 13.1 Ma [112]. These data suggest the presence of a long undocumented gap in the fossil record of Southeast Asia, since no remains assigned to the tribe Bovini have been described in this region before the Pleistocene.
4.8 Development of modern terrestrial fauna during the Late Miocene and Plio-Pleistocene
Our molecular timescale shows that most of the current biodiversity of bovid and cervid tribes took place during the last 12 million years. Their recent diversification may be explained by three main factors: (1) the expansion of grasslands, (2) the presence of biogeographic corridors, which enabled faunal dispersals between Africa and Eurasia, Eurasia and North America, and North America and South America, and (3) the contemporaneous radiation of large carnivores, such as Felidae, Canidae, and Hyaenidae, which are the main predators of bovids and cervids.
Particularly spectacular is the evolutionary history of gazelles, which was marked by three successive phases of rapid diversification during the late Neogene. The first phase of radiation began at the Middle Miocene/Late Miocene boundary, i.e., between 12.0 ± 1.6 and 11.0 ± 1.5 Ma, when the tribe Antilopini split into four lineages, including the Asian subtribe Procaprina (Procapra), and three African subtribes, i.e., Antilopina (gazelle-like genera), Raphicerina (Raphicerus, Dorcatragus, Madoqua) and Ourebina (Ourebia). This period coincides with the divergence between the three tribes Alcelaphini, Caprini, and Hippotragini, between 12.5 ± 1.8 Ma and 12.0 ± 1.9 Ma. Interestingly, all bovid species adapted to desert environments come from the tribes Antilopini (e.g., Gazella leptoceros), Caprini (e.g., Ammotragus lervia) and Hippotragini (e.g., Addax nasomaculatus). There is little doubt, therefore, that their diversification was promoted by the development of more open habitats during the late Neogene. Fossil plants indicate that the earliest grasslands/savanna appeared in East Africa in the Mid-Miocene [120,121]. In addition, the global vegetation reconstruction recently provided by Pound et al. [122] for the Tortonian (11.61–7.25 Ma) suggests that tropical xerophytic shrubland was the dominant biome in northern Africa, East Africa, and in the Arabian Peninsula. All these data are congruent with the hypothesis that the first radiation of Antilopini and that of the Alcelaphini–Caprini–Hippotragini clade took place in the northern and eastern parts of Africa, which may have facilitated their dispersals in Eurasia. The second radiation of Antilopini occurred between 8.5 and 8.0 ± 1.3 Ma, when the subtribe Antilopina diversified into three lineages, corresponding to the Asian genus Saiga, true gazelles, and an African clade containing Antidorcas and Litocranius, as well as Ammodorcas (not represented here, but see Ropiquet et al. [26]). That epoch can be linked to the global expansion of C4 grasslands between 8.5 and 6.5 Ma [123], which favored the diversification of most bovid tribes during the Late Miocene [113]. The third radiation of Antilopini occurred during the early Pliocene, when the four genera of gazelles (Gazella, Eudorcas, Nanger, and the Indian genus Antilope) diverged from each other between 4.5 and 4.2 ± 0.9 Ma. During this time, both North and East Africa seem to have experienced a drier climate leading to conditions more like those seen today [124,125]. The fact that one Asian lineage emerged from each of the three successive radiations of Antilopini implies that all these phases of diversification were possibly accompanied by faunal migration from Africa to Eurasia.
The evolution of South American deer is another interesting case of radiation. This radiation is traditionally explained by the arrival of their common ancestor in South America at 3–3.5 Ma, i.e., after the formation of the Isthmus of Panama [39,126]. As in Gilbert et al. [39], our molecular analyses suggest, however, that the Pliocene evolution of American deer was more complex. Our phylogenetic results show that South American deer fall into two clades: one includes only South American deer, and another one contains both South and North American deer. Molecular estimates indicate that the common ancestor of the South American clade underwent a rapid radiation between 3.4 and 3.0 ± 0.4 Ma, i.e., just after the completion of the Pliocene land bridge. Our interpretation is that American deer diversified firstly in the northern subcontinent, as at least two lineages were already differentiated at 5.1 ± 0.5 Ma. This hypothesis is supported by the fossil record of North America, which shows the existence of two divergent odocoileine lineages at the end of Miocene [30].
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgments
AH and AR would like to dedicate this paper to the memory of our friend and colleague, the late Annie Tillier. We are very grateful to people who provided tissue samples: Junghwa An, Eric Bonnem, Jean-Marie Carenton, Arnaud Delapre, Norin Chai, Bertrand Chardonnet, Philippe Chardonnet, Bertrand Des Clers, Raphaël Cornette, Gauthier Dobigny, Emmanuel Douzery, Bernard Dutrillaux, Céline Canler, Jim Heffelfinger, Françoise Hergueta-Claro, Jean-Pierre Hugot, Aude Lalis, Sibyle Moulin, Therry Oberdorff, Link E. Olson, Stéphane Ostrowski, Claire Rejaud, Jacques Rigoulet, Roland Simon, Theerapol Sirinarumitr, Alexander Sliwa, Christophe Voisin, Vitaly Volobouev. AH would like to thank Giovanni Bianucci, Jean-Renaud Boisserie, and Christian de Muizon for bibliography. This is contribution ISEM 2011-155 of the Institut des Sciences de l’Évolution de Montpellier. This work was supported by the MNHN, CNRS, PPF “Etat et structure phylogénétique de la biodiversity actuelle et fossile”, “Consortium National de Recherche en Génomique”, and The World Agroforestry Centre, Sida. It is part of agreement No. 2005/67 between Genoscope and MNHN on the project “Macrophylogeny of life”.