

1 Introduction
Chelate complexes obtained from diphosphanes where the phosphorus centres are separated by more than twenty bonds remain relatively uncommon 〚1–3〛. Reaction of such large bidentate ligands with transition metal complexes often produces oligomeric material rather than chelate structures 〚4, 5〛. As already noted by Ogino, the chemical literature contains many examples of supposed chelate complexes, but for many of them the ring structure has not been firmly established 〚6〛. This concerns in particular ligands that contain more than three or four bonds between the coordination centres. A rational way to favour chelating over bridging behaviour consists in performing the complexation reaction at high dilution, but this requires the coordination steps to proceed quickly. Alternatively, preorganisation of the complexing units brought about by increasing the ligand backbone rigidity may result in better metal chelation. We have recently studied the coordinative behaviour of a series of large bidentate ligands built on bucket-shaped units, notably cyclodextrin and calixarene backbones. All these ligands consisted of two podands tethered at the same rim of a cone-shaped cavity. We found that such long chain diphosphines systematically lead to chelate complexes when opposed to cationic species bearing labile ligands, e.g. 〚Rh(COD)(solvent)2〛+, 〚PdMe(solvent)2(COD)〛+, 〚Ru(p-cymene)(solvent)3〛2+, 〚Au(solvent)2〛+, etc., whilst their reaction with neutral starting complexes containing poorer leaving groups (e.g. MeCN) usually affords oligomeric compounds 〚7, 8〛.
Although a number of studies have been carried out on P(III)-derivatised CDs, none deals with phosphite ligands 〚1, 9–17〛. As part of our studies on cavity-derived bidentates, we report here on the synthesis of diphosphites L1 and L2 (Fig. 1), both built on an α-cyclodextrin platform, and their ability to form catalytically active large chelate complexes with transition metals. In the following, the letters ‘a’ and ‘b’ refer to AD- and AC-disubstituted CDs, respectively. A unique example of an α-CD capped with a short bridging fragment is also reported.
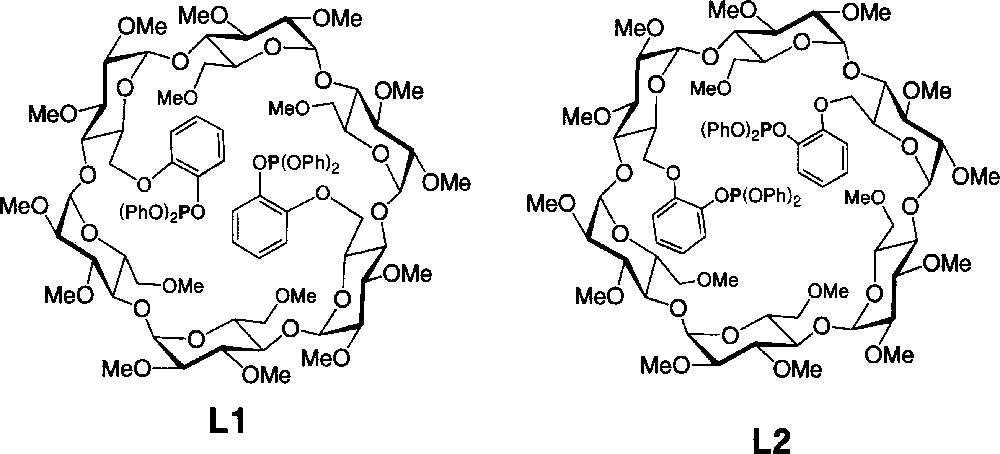
Diphosphites L1 and L2.
2 Results and discussion
2.1 Syntheses of ligands and complexes
The C2-symmetrical ligand L1 was synthesised in three steps from the known dimesylate 1a (Fig. 2) 〚18〛. Thus, reaction of 5 equiv of sodium 2-(benzyloxy)phenolate with 1 in DMF at 70 °C afforded a mixture of three products among which we isolated the desired compound 2a in 65% yield. The presence of three doublets for the H-1 protons (see experimental part) as well as eight singlets for the methyl groups in the 1H NMR spectrum is consistent with a molecule having C2 symmetry, thus confirming substitution of both available mesylate groups. Partial loss of one benzyl group under these reaction conditions led to the formation of 3a (7%). Its 1H NMR spectrum reveals the absence of symmetry and indicates clearly the presence of a phenolic proton. The 1H NMR spectrum of the third species 4a (17%) is also indicative of C2 symmetry. However, compared to 2a, the H-1 signals of 4a are much more spread out (Δδ = 0.30 ppm vs 0.11 ppm for 2a), indicating the presence of a distorted CD matrix (Fig. 3). As confirmed by a single crystal X-ray diffraction study (vide infra), the latter results from the capping of the CD platform with a short 1,2-phenylene unit, characterised by a four-proton AA’BB’ system in the aromatic region of the 1H NMR spectrum. Only benzyl cleavage of the monosubstituted intermediate 5a (Fig. 4) and subsequent intramolecular substitution of the remaining mesylate group can explain the formation of such a capped derivative. In the solid state (Figs. 5 and 6), the C(11) and C(21) glucose units and their symmetrical counterparts are strongly tilted with respect to the plane constituted by the six O-4 atoms (tilt angles: 35.0(3)° (C(11)) and 31.8(3)° (C(21)), vs 14.4(3)° for the C(1) ring), so as to accommodate the capping unit (the tilt angle is defined by the dihedral angle between the best plane defined by the six O-4 atoms and the plane defined by the C-1, C-4, O-4 and O-4’ atoms of a particular glucose unit). Remarkably, in spite of the considerable strain imposed on the CD framework 〚19〛, all glucose units display the usual 4C1 conformation 〚20〛. The 1,2-phenylene moiety, which adopts two possible orientations (occupancy 1:1), is inclined by ± 39.5° with respect to the CD axis. Note that the 2D NMR ROESY spectrum contains cross-peaks, corresponding to through-space interactions between the phenylene and all OMe-6 protons. In keeping with the fact that the molecule has averaged C2-symmetry, this observation indicates a fan-like motion of the phenylene ring above the CD cavity in solution. The same capped species was obtained in moderate yield (ca 30%) when the dimesylate was reacted with disodium catecholate. However, in this case, some oligomeric material was formed. Treatment of 2a and 3a with H2 in the presence of Pd/C afforded the diphenol 6a quantitatively. The latter reacted smoothly with one equivalent of chlorodiphenylphosphite ClP(OPh)2 in the presence of NEt3 at –40 °C to give ligand L1, whose 31P NMR spectrum consists of a singlet at 127.5 ppm, commensurate with the expected C2 symmetry. In L1, 27 bonds separate the two phosphorus atoms.
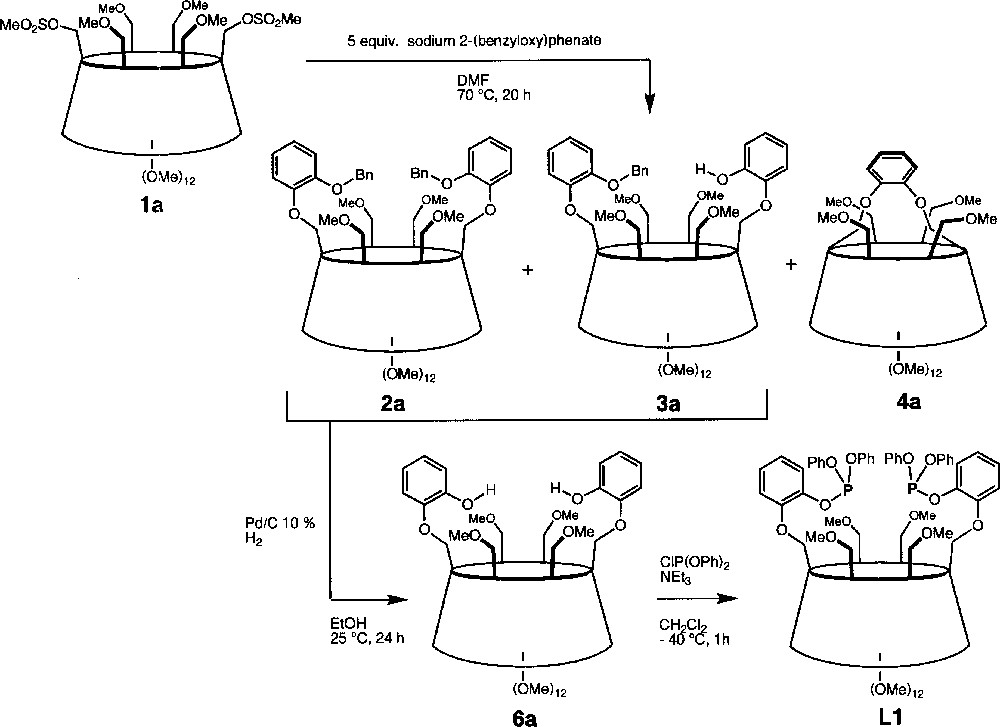
Synthesis of the C2-symmetrical ligand L1 from the dimesylate 1a.
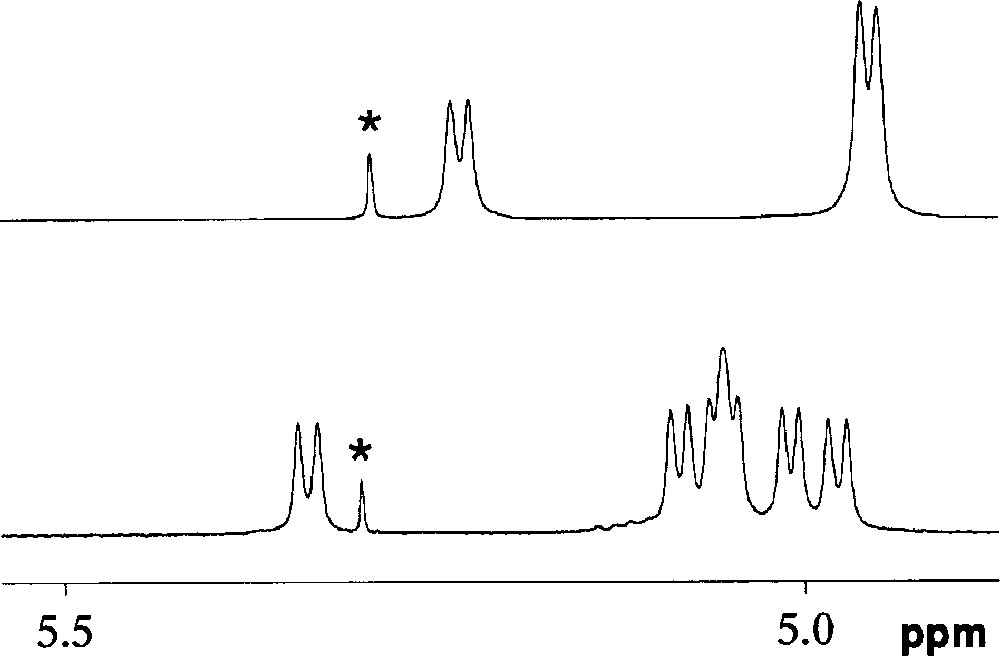
Part of the 1HNMR spectra (CDCl3) of 4a and 4b showing the dispersion of the anomeric H-1 signals. The peak marked with an asterisk is due to residual CH2Cl2.
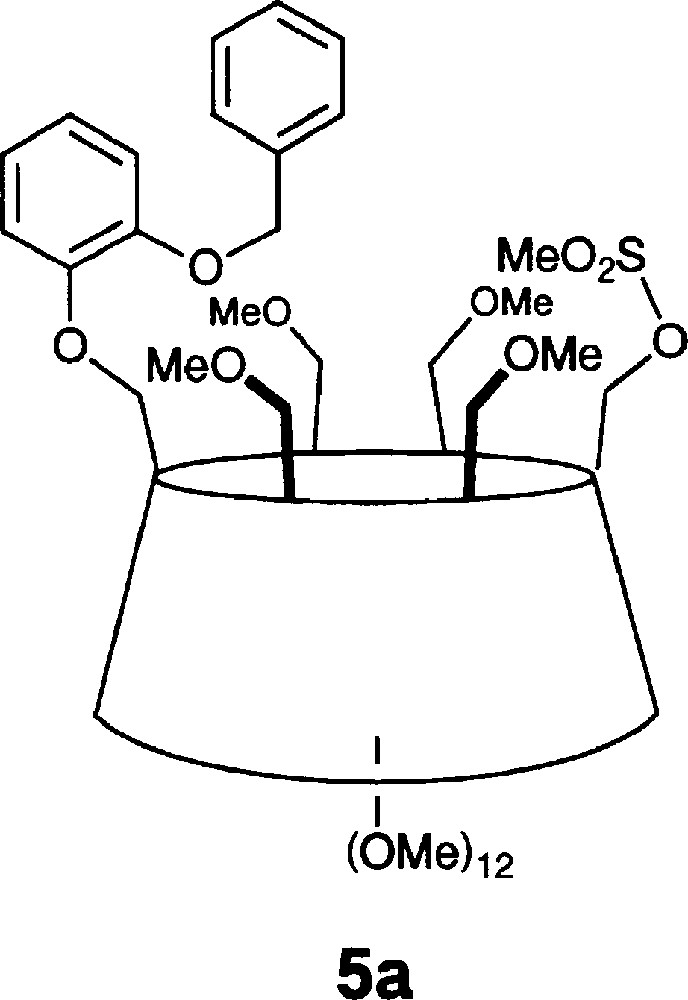
Compound 5a.
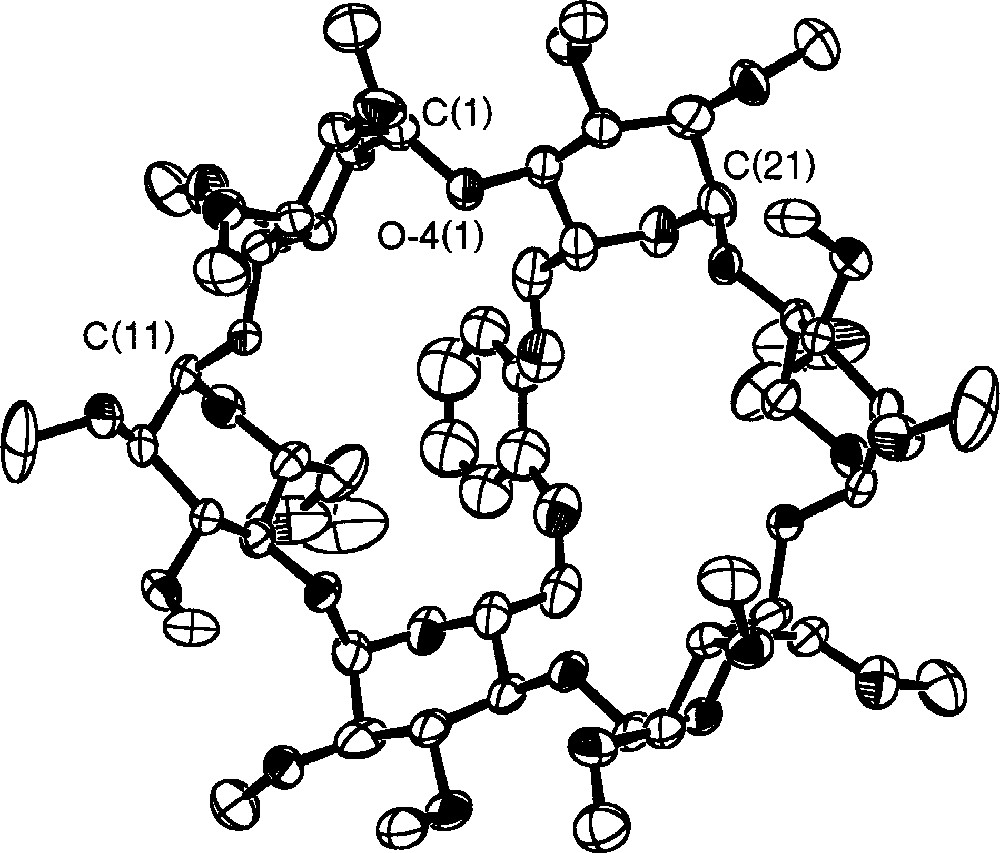
View of the 1,2-phenylene-capped CD 4a along the C2 axis (the bridging unit lies in the backside). Only one orientation of the catecholate is shown. The included heptane molecule has been omitted for clarity.
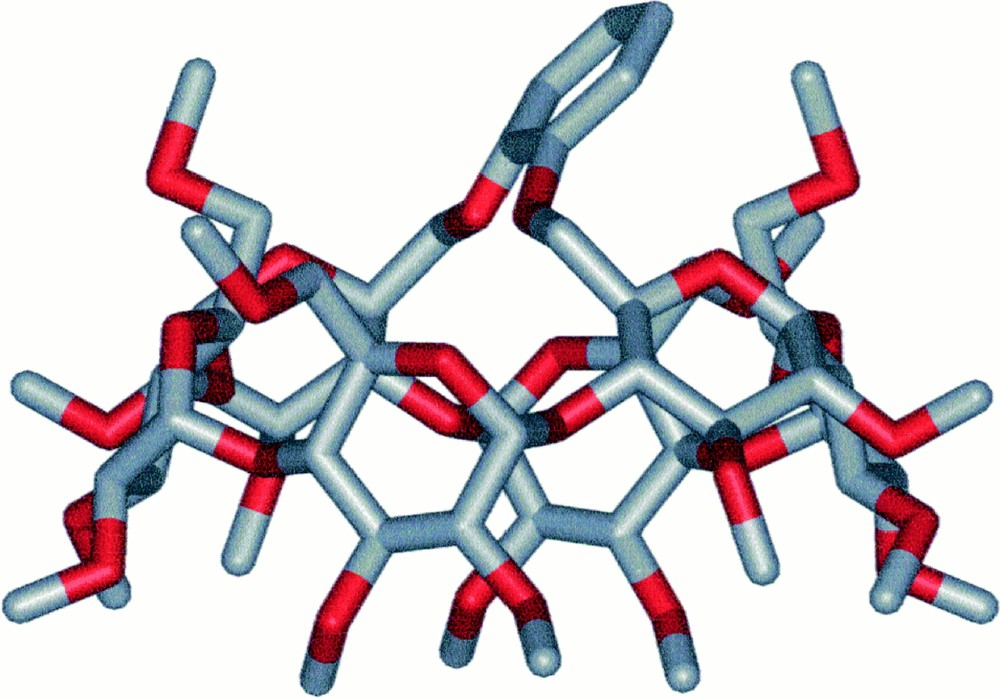
Side-view of 4a (sticks).
The bis(triaryl)phosphite regioisomer L2 was synthesised from the known tris(4-tert-butylphenyl)methyl AC-disubstituted CD derivative 7b. Thus, removal of the protecting groups was achieved with aqueous HBF4 in MeCN to give diol 8b, which was then converted to dimesylate 1b using MsCl in pyridine in the presence of 4-(dimethylamino)pyridine (DMAP) (Fig. 7). As for 1a, treatment of 1b with 5 equiv of sodium 2-(benzyloxy)phenolate produced a mixture of four products, amongst which the desired disubstituted product 2b was isolated in 57% yield. Again, bridging of two primary positions with a catechol unit was found to take place and the capped species 4b was isolated in 18% yield. As for 4a, the large dispersion of the H-1 signals (Δδ = 0.37 ppm vs 0.11 ppm for 2b) in the 1H NMR spectrum indicates the presence of a distorted CD torus (Fig. 3). However, as revealed by 2D ROESY experiments, the phenylene protons are in close proximity to only three of the four OMe-6 protons, suggesting that the phenylene plane is tilted towards the cavity centre and does not swing in this case about the O-6A—O-6C axis. Again, two inseparable species whose 1H NMR spectra are consistent (see experimental part) with products resulting from cleavage of one benzyl group from 2b were also obtained in very low yield. Catalytic hydrogenation of 2b afforded diphenol 6b quantitatively, the 1H and 13C NMR spectra of which show remarkable asymmetry. The latter may result from the presence of hydrogen bonding between the two hydroxy groups and the CD matrix, which helps rigidifying the overall structure. Compound 6b reacted smoothly with 2 equiv of ClP(OPh)2 to give ligand L2. Surprisingly, both phosphorus atoms appear as magnetically equivalent (δ 127.5 ppm) in the 31P NMR spectrum.
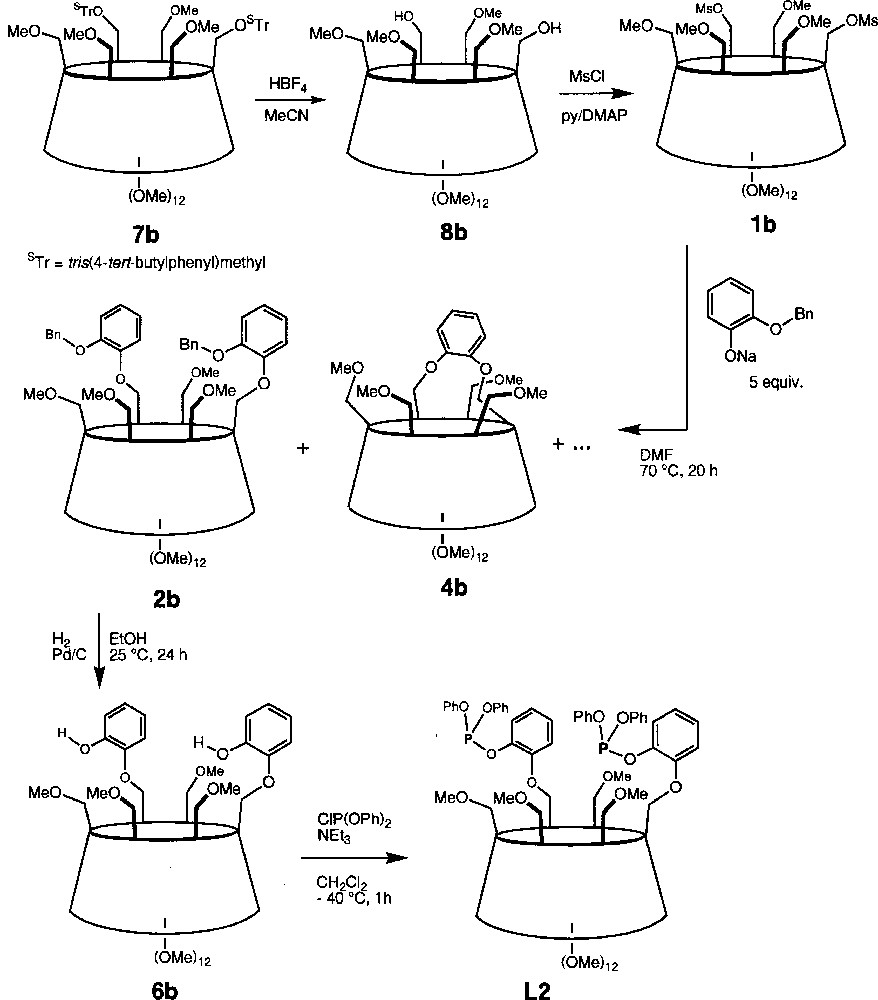
Chemical route from 7b to L2.
The bimetallic complexes 9a and 9b (Fig. 8) were readily obtained in high yields by reacting 〚o-C6H4CH2NMe2)PdCl〛2 with the corresponding diphosphite. Both complexes are characterised by a singlet at ca 104.5 ppm in the 31P NMR spectrum. Note that for 9b complexation does not result in the differentiation of the two formally non-equivalent phosphorus atoms. Coordination of both phosphorus-binding sites was confirmed by the presence of 4J(PH) coupling constants (3.3 Hz 〚2 ×〛 in 9a and 3.3 and 4.9 Hz in 9b) between NMe protons and the trans-bonded phosphorus atom. The 1H NMR spectra of both 9a and 9b show that the two methyl groups carried by each nitrogen atom are magnetically non-equivalent, in accord with the chirality of these molecules.
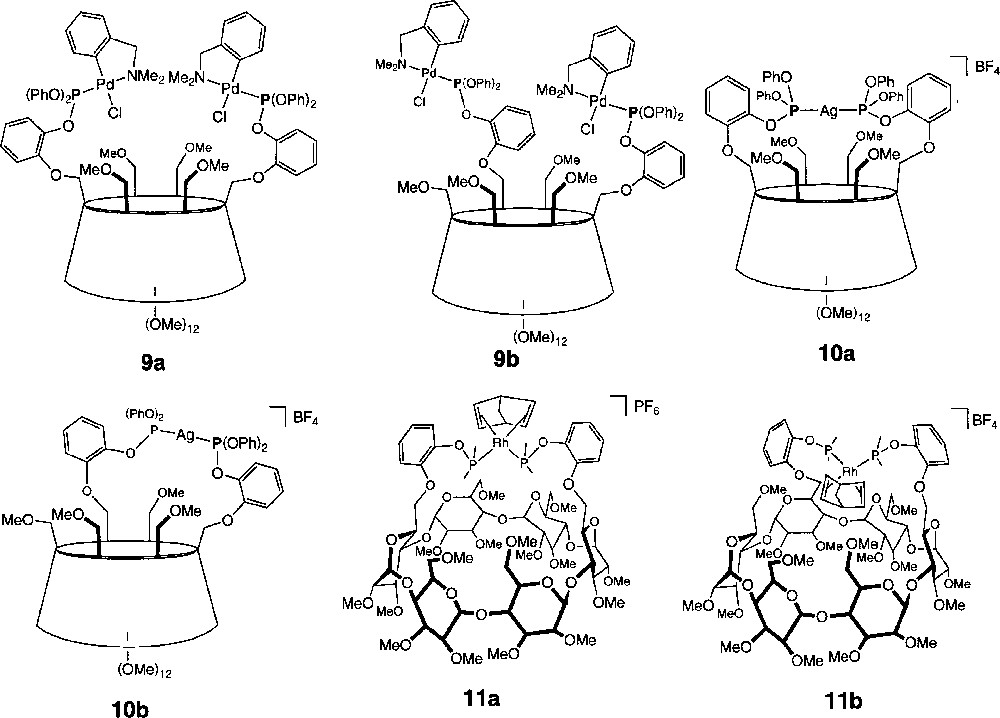
Compounds 9a, 9b, 10a, 10b, 11a and 11c.
When either L1 or L2 was treated with 1 equiv AgBF4, the exclusive formation of the chelates 10a/10b was observed (Fig. 8). The capping of the CD cavity with an Ag+ cation was confirmed by FAB–MS spectrometry. Both complexes produce an intense peak at 1921.3, corresponding to the molecular ion (M+–BF4). As expected, in the 31P NMR spectrum of C2-symmetrical 10a, the phosphorus atoms give rise to two doublets centred at 116.7 ppm (J(107AgP) = 1083 Hz, J(109AgP) = 1231 Hz), whereas they resonate as two ABX systems (X = 107Ag and 109Ag, δA = 108.3 and δB = 114.5 ppm, 2J(PP’) = 256 Hz, J(107AgP’) = 977 Hz, J(109AgP’) = 1088 Hz) in 10b. The J(AgP) values are rather large when compared to those of phosphine silver complexes, but this has also been observed recently for the J(PtP) coupling constants of tris(aryl)phosphite platinum(II) complexes 〚21〛. Unlike that of 10a, the 1H NMR spectrum of 10b is somewhat broad, which suggests fluxional behaviour, possibly resulting from exchanging methoxy groups that compete for coordination to the silver cation. Similar dynamics have been observed recently in other silver-capped CDs 〚9〛. Likewise, chelate complexes 11a/11b were obtained in high yields when the in situ formed cationic precursors 〚Rh(NBD)(THF)2〛PF6 and 〚Rh(NBD)(THF)2〛BF4 were reacted with one equivalent of L1 and L2, respectively. Once again, the FAB-MS spectra of 11a/11b (Fig. 8) left no doubt on the monomeric nature of the complexes, showing intense peaks at 2007.7 and 2008.4 for the 〚M–PF6〛+ (11a) and 〚M–BF4〛+ (11b) cations, respectively. Both 31P NMR spectra display a single doublet centred at 110.5 ppm (J(RhP) = 264 Hz (11a), 259 Hz (11b)). It is interesting to point out that again the phosphorus atoms of 11b give rise to an A2X pattern (X = Rh), despite the complex lack of symmetry. This unexpected feature arises probably from the fact that the distance separating the phosphorus atoms from the chiral cavity is quite large in the absence of MeO-6 ether coordination. Furthermore, at room temperature, the 1H NMR spectra of 11a and 11b are both broad, suggesting that these molecules are dynamic in solution. Recording the 1H NMR spectrum of 11a at 60 °C caused all signals to sharpen, but did not reduce their number. These findings suggest that the metal plane is tilted with respect to the CD axis and that the metal-containing handle is subject to conformational changes. No indication for a metal plane describing a pendulum motion about the P,P axis could be inferred from our observations. Finally, we observed that reaction of L1 or L2 with 〚Rh(COD)2〛BF4, which contains poorer leaving ligands, produces a mixture of monomeric and oligomeric species. Undoubtedly, the selective formation of chelate complexes using 〚Rh(NBD)(THF)2〛+ originates from a combination of the following features: firstly, the use of starting cationic complexes bearing highly labile ligands promotes a fast coordination of both phosphorus centres. Secondly, the rigid CD core orientates the two phosphite substituents in the same direction and therefore maintains the two phosphorus centres in close proximity.
2.2 Catalytic properties of L1 and L2
Our catalytic attempts focused on the hydroformylation of 1-octene and the hydrogenation of prochiral dimethyl itaconate, two commonly used reactions for testing P(III) ligands 〚22〛. For the hydroformylation studies, the complexes were formed in situ according to standard literature procedures, using 〚Rh(CO)2(acac)〛 as starting complex. Thus formation of complexes other than chelate ones cannot be excluded.
2.2.1 Hydroformylation
The catalytic performances of L1 and L2 are summarised in Table 1. Both rates and regioselectivities are comparable to those observed with the flexible and bulky diphosphite 12 (Fig. 9; l/b = 2.2, TOF = 1550, isomerisation 20%) but the percentage of isomerisation does not exceed 5% in our case 〚23〛. Consistent with van Leeuwen’s observations made for other diphosphites, the flexible nature of our chelating complexes probably accounts for the observed poor linear aldehyde selectivity. Interestingly, activity is doubled on going from L1 to L2 (entries 3 and 4), probably reflecting a larger steric hindrance about the metal centre in the L2/rhodium system. This feature is likely to reduce the number of coordinated phosphorus atoms during the catalytic cycle, which in turn leads to less stable hence more reactive intermediate species. Raising the octene/rhodium ratio from 600 to 1200 produced a two-fold increase of the reaction rate. Such a substrate concentration dependency has also been observed by van Leeuwen when operating in low olefin concentration 〚22, 23〛. No evidence was found for a supramolecular interaction between the CD cavity and the olefinic substrate under the used reaction conditions (solvent: toluene).
Hydroformylation of 1-octene.
| Entry | Ligand | L/Rh ratio | S/Rh ratio | % Conv.a (1 h) | Aldehyde selectivity (%) | l/b ratio | TOFb |
| 1 | L1 | 5 | 600 | 69 | 93 | 1.8 | 300 |
| 2 | L1 | 5 | 1200 | 56 | 91 | 1.9 | 550 |
| 3 | L2 | 5 | 600 | 98 | 95 | 1.9 | 600 |
| 4 | L2 | 5 | 1200 | 98 | 93 | 2.0 | 1200 |
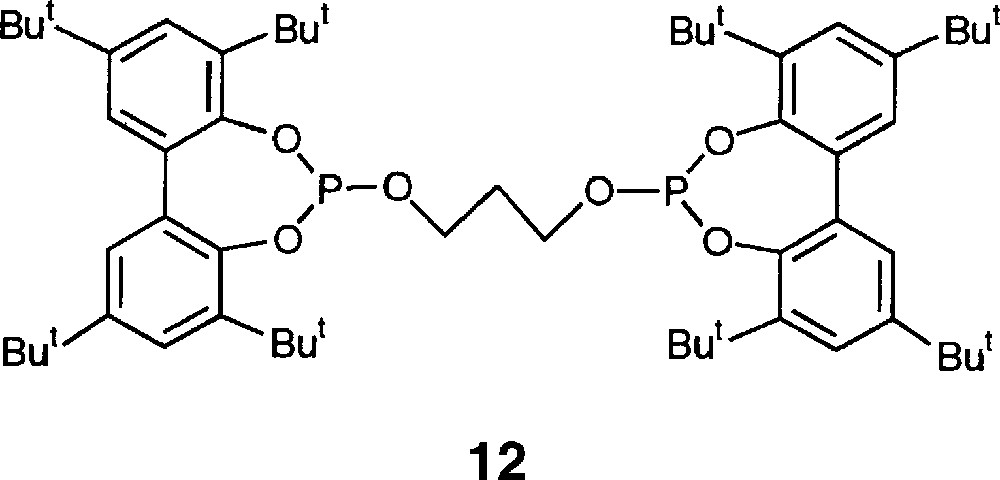
Compound 12.
2.2.2 Hydrogenation
In our first experiments, the hydrogenation catalysts were formed in situ by reacting 〚Rh(COD)2〛BF4 with either L1 or L2 in CH2Cl2 solution before adding the substrate. As described previously, under these conditions mixtures of monomeric and oligomeric compounds are formed. Both systems exhibit moderate activity (Table 2, entries 1 and 2). With EEs not exceeding 17%, the observed enantioselectivities are rather disappointing. We then repeated the tests with the preformed chelate complexes 11a and 11b (entries 3 and 4). To our surprise, only 11b proved to be an active hydrogenation catalyst, whereas 11a showed very poor activity. In addition, 11b produces remarkable enantioselectivity (EE 83.6%, R isomer). Clearly, access to the metal centre is facilitated in the unsymmetrical AC-capped system, but remains below that of the less crowded oligomeric species formed when the catalysts are formed in situ. It is plausible that with complex 11b, the chiral cavity that is rigidly positioned near the metal centre favours entrapment and orientation of the olefinic substrate, so as to induce good enantioselectivity.
Hydrogenation of dimethyl itaconate.
| Entry | Catalyst | S/Rh ratio | % Conv. (24 h)a | EE | TOFb |
| 1 | 〚Rh(COD)2〛BF4/L1c | 500 | 58 | 10 | 13 |
| 2 | 〚Rh(COD)2〛BF4/L2c | 500 | 47 | 17 | 10 |
| 3 | 11ad | 500 | 1 | n. d.e | 0.2 |
| 4 | 11bd | 500 | 17 | 83.6 | 3.5 |
3 Conclusion
In this paper, we have shown that by grafting two phosphite ligands onto a methylated CD platform at either AC or AD positions, diphosphites with chelating properties can be obtained. Metallomacrocycles consisting of up to 29 membered-rings were produced as the sole products when the preorganised diphosphites were reacted with transition metal cations bearing labile ligands. The interesting hydrogenation properties displayed by complex 11b illustrate the potential of unsymmetrically capped CDs as enantioselective catalysts.
4 Experimental section
4.1 General procedures
All commercial reagents were used as supplied. All manipulations involving phosphines were performed in Schlenk-type flasks under argon. Solvents were dried by conventional methods and distilled immediately prior to use. Column chromatography was performed on silica gel 60 (particle size 40–63 μm, 230–240 mesh). CDCl3 was passed down a 5 cm-thick alumina column and stored under argon over molecular sieves (4 Å). Routine 1H, 31P{1H}, and 13C{1H} NMR spectra were recorded with FT Bruker AC200 and AC300 instruments at 25 °C, while 500-MHz spectra were recorded on an ARX 500 Bruker instrument. 1H NMR spectral data were referenced to residual protiated solvents (7.26 for CDCl3), 13C chemical shifts are reported relative to deuterated solvents (77.0 ppm for CDCl3), and the 31P NMR data are given relative to external H3PO4. All mass spectra were recorded on a ZAB HF VG Analytical spectrometer using m-nitrobenzyl alcohol as matrix. Cyclodextrins 1a 〚18〛 and 7b 〚24〛, chlorodiphenylphosphite 〚25, 26〛 and complexes 〚(o-C6H4CH2NMe2)PdCl〛2 〚27〛, 〚RhCl(NBD)〛2 〚28〛 and 〚Rh(COD)2〛BF4 〚13〛 were synthesised according to literature procedures. DMBA stands for o-dimethylbenzylamine. The numbering of the atoms within a glucose unit is as follows:
Hydroformylation studies were performed in a stainless steel autoclave (100 ml) containing a glass beaker and a magnetic stirring bar. The beaker was charged with toluene solutions of both the diphosphite ligand and 〚Rh(CO)2(acac)〛 (total volume 20 ml), followed by the internal standard (decane). The autoclave was flushed several times with CO/H2 (1:1, v/v), then pressurized to 20 bar, and finally heated to 80 °C for 1 h. After depressurising and cooling down to room temperature, the substrate (1-octene) was added, and the mixture was again pressurised and heated to reaction temperature. Samples were taken at different reaction times and analysed by GC.
Hydrogenation experiments were carried out in a Schlenk tube under ambient pressure and room temperature. Experiments using 〚Rh(COD)2〛BF4: the diphosphite ligand was dissolved in 15 ml of CH2Cl2. After addition of 〚Rh(COD)2〛BF4, the solution was saturated with H2 for 10 min, whereupon the substrate (dimethyl itaconate) was added. Samples were taken at different reaction times and analysed by GC. Enantiomeric excess was determined by 1H NMR using an optically active shift reagent (tris 〚3-heptafluoropropylhydroxymethylene)-(+)-camphorato〛 europium (III) ((+)-Eu(hfc)3)*. A CDCl3 solution of the organic substrate was prepared in an NMR tube and small amounts of the shift reagent were gradually added until a clean splitting of the peaks was achieved (ca 5% of shift reagent). Experiments using 11a or 11b: the complex was dissolved in 15 ml of CH2Cl2, and the solution was saturated with H2 for 10 min. Then dimethyl itaconate was added. The reaction was followed by GC. The enantiomeric excess was determined by GC using a chiral column.
4.2 Syntheses
4.2.1 Synthesis of 2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin (8b)
Tetrafluoroboric acid (34%, 24.1 ml) was added to a solution of 7b (12.000 g, 5.95 mmol) in MeCN (50 ml). The solution was stirred for 20 min at room temperature whereupon Et3N (45 ml) was added. Addition of water (1000 ml) caused sTrOH to precipitate. The latter was filtered and the filtrate extracted with CH2Cl2 (4 × 300 ml). The organic extract was washed with saturated aqueous NaHCO3 (2 × 300 ml) before being dried (MgSO4), and evaporated to dryness. The residue was subjected to column chromatography (SiO2, CH2Cl2/MeOH 88:12,v/v) to afford 8b as a colourless solid (6.04 g, 85%). Rf (CH2Cl2/MeOH, 9 : 1, v/v) = 0.32; Mp 162–164 °C; 1H NMR (200 MHz, C6D6): δ = 3.29 (s, 6H, OCH3), 3.30 (s, 6H, OCH3), 3.32 (s, 3H, OCH3), 3.33 (s, 3H, OCH3), 3.37 (s, 3H, OCH3), 3.38 (s, 3H, OCH3), 3.39 (s, 3H, OCH3), 3.41 (s, 6H, OCH3), 3.74 (s, 3H, OCH3), 3.75 (s, 12H, OCH3), 3.76 (s, 3H, OCH3), 3.09–4.35 (36H, H-2, H-3, H-4, H-5, H-6), 5.01 (d, 3JH-2,H-1 = 3.5 Hz, 1H, H-1), 5.04–5.08 (overlapping signals, 3H, H-1), 5.11–5.12 (overlapping signals, 2H, H-1); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.76, 57.82 〚× 2〛, 57.92, 58.81, 58.87 and 58.97 〚× 4〛 (CH3O-2, CH3O-6), 61.50 〚× 2〛 and 61.63 〚×4〛 (CH3O-3), 62.15, 62.20, 71.30, 71.39, 71.46, 71.62 (C-6), 71.10 〚× 2〛, 71.17 〚× 2〛, 72.33, 72.54 (C-5), 81.23 〚× 6〛, 81.72, 81.95 〚× 5〛, 82.11 〚×5〛, 82.24 (C-2, C-3, C-4), 99.32, 99.52 and 99.75 〚× 4〛 (C-1). Anal. calcd for C52H92O30·0.5 CH2Cl2 (1197.27 + 42.47): C 50.86, H 7.56; found: C 50.63, H 7.93.
4.2.2 Synthesis of 6A,6C-di-O-methylsulfonyl-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B, 6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin (1b)
Methylsulfonyl chloride (1.180 g, 10.29 mmol) was added to a solution of 8b (5.600 g, 4.68 mmol) and N,N-dimethylpyridine (0.930 g, 7.58 mmol) in anhydrous pyridine (80 ml). The reaction mixture was stirred overnight at room temperature whereupon brine (150 ml) was added. The solution was then extracted with EtOAc (4 × 200 ml), and the organic phase washed respectively with HCl 2M (2 × 200 ml), NaOH 2 M (200 ml), before being dried (MgSO4) and filtered. Removal of the solvent in vacuo afforded pure 1b as a colourless solid. Rf (CH2Cl2/MeOH, 9 : 1, v/v) = 0.44; Mp 113–115 °C; 1H NMR (200 MHz, C6D6): δ = 2.82 (s, 3H, OSO2CH3), 2.84 (s, 3H, OSO2CH3), 3.26 (s, 3H, OCH3), 3.28 (s, 9H, OCH3), 3.31 (s, 6H, OCH3), 3.38 (s, 3H, OCH3), 3.41 (s, 3H, OCH3), 3.43 (s, 3H, OCH3), 3.45 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 3.69 (s, 6H, OCH3), 3.71 (s, 6H, OCH3), 3.76 (s, 3H, OCH3), 3.06–4.45 (34H, H-2, H-3, H-4, H-5, H-6), 4.76 (dd, 3JH-5,H-6a = 4.5 Hz, 2JH-6b,H-6a = 11.3 Hz, 1H, H-6), 4.92 (dd, 2JH-6a,H-6b = 11.3 Hz, 1H, H-6), 4.96 (d, 3JH-2,H-1 = 3.7 Hz, 1H, H-1), 5.00 (2d, 3JH-2,H-1 = 3.7 Hz, 2H, H-1), 5.02 (d, 3JH-2,H-1 = 3.9 Hz, 1H, H-1), 5.15 (2d, 3JH-2,H-1 = 3.4 Hz, 2H, H-1); 13C{1H} NMR (50 MHz, CDCl3): δ = 37.26 and 37.48 (OSO2CH3), 57.56, 57.63, 57.69, 58.02, 58.38, 58.81 〚× 2〛, 59.00 〚× 3〛 (CH3O-2, CH3O-6), 61.43, 61.59 〚× 3〛, 61.69 and 61.76 (CH3O-3), 69.53, 70.94, 71.00 〚× 2〛, 71.20, 71.33 (C-5), 69.92 〚× 3〛, 70.08, 71.53, 71.85 (C-6), 80.84 〚× 2〛, 80.95 〚× 2〛, 81.03, 81.16, 81.33, 81.49, 81.75, 81.88 〚× 2〛, 81.98, 82.11 〚× 2〛, 82.24 〚× 2〛, 82.31, 83.13 (C-2, C-3, C-4), 98.60, 98.90, 99.98, 100.21 〚× 2〛 and 100.40 (C-1). Anal. calcd for C54H96O34S2·0.5 CHCl3 (1353.45 + 59.69): C 46.32, H 6.88; found: C 46.28, H 7.08.
4.2.3 Syntheses of 6A,6D-bis-O-(2-benzyloxyphenyl)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin (2a) and 6A,6C-bis-O-(2-benzyloxyphenyl)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F- hexadeca-O-methyl-α-cyclodextrin (2b)
NaH (60%, 0.170 g, 4.29 mmol) was added to a solution of 2-benzyloxyphenol (0.860 g, 4.29 mmol, 0.98 ml) in DMF (25 ml). After 20 min, 1a (1.158 g, 0.86 mmol) was added. The solution was stirred overnight whereupon water (50 ml) was added. The mixture was extracted with Et2O (4 × 100 ml), and the ether phase was subsequently washed with an aqueous 2M NaOH solution (100 ml), dried over MgSO4 and filtered. Removal of the solvent in vacuo gave a colourless residue (1.832 g), which was purified by column chromatography (SiO2, hexane/ethyl acetate 20 : 80 → 0: 100, v/v) to afford three fractions:
Fraction 1. 6A,6D-di-O-(2-benzyloxyphenyl)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin (2a) (0.869 g, 65%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.37; Mp 100–102 °C; 1H NMR (200 MHz, CDCl3): δ = 3.23 (s, 6H, OCH3), 3.31 (s, 6H, OCH3), 3.37 (s, 6H, OCH3), 3.47 (s, 6H, OCH3), 3.51 (s, 6H, OCH3), 3.63 (s, 6H, OCH3), 3.66 (s, 12H, OCH3), 2.94–4.23 (32H, H-2, H-3, H-4, H-5, H-6B,C,E,F), 4.31 (d, 2JH-6b,H-6a = 10.1 Hz, 2H, H-6aA,D), 4.57 (dd, 3J= 3.2 Hz, 2JH-6b,H-6a = 10.1 Hz, 2H, H-6bA,D), 5.02 (d, 3JH-1,H-2 = 3.2 Hz, 2H, H-1A,D), 5.08 (t, 4J = 1.7 Hz, 4H, OCH2Ph), 5.12 (2d, 3JH-1,H-2 = 3.2 Hz, 4H, H-1B,C,E,F), 6.84–6.99 (8H, catecholate H), 7.24–7.44 (10H, benzyl arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.52, 57.92, 57.95, 58.93, 59.10 (CH3O-2, CH3O-6), 61.79, 61.85 〚× 2〛 (CH3O-3), 68.64 (OCH2Ph), 70.90 (C-6A,D), 71.13,71.39 〚× 2〛 (C-5), 71.49 〚× 2〛 (C-6B,C,E,F), 81.29 〚× 3〛, 82.14 〚× 2〛, 82.27, 82.50 〚× 2〛, 82.90 (C-2, C-3, C-4), 99.84 (C-1A,D), 100.20, 100.27 (C-1B,C,E,F), 114.86, 115.15, 121.41, 121.58 (catecholate CH), 126.8, 127.67, 128.50 (benzyl arom. CH), 137.58, 149.04, 149.41 (Cipso). Anal. calcd for C78H112O32 (1561,71): C 59.99, H 7.23; found: C 59.94, H 7.19.
Fraction 2. 6A-O-(2-benzyloxyphenyl),6D-O-(2-hydroxyphenyl)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin (3a) (0.086 g, 7%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.23; Mp 123–126 °C; 1H NMR (200 MHz, CDCl3): δ = 3.19 (s, 3H, OCH3), 3.21 (s, 3H, OCH3), 3.26 (s, 3H, OCH3), 3.34 (s, 3H, OCH3), 3.38 (s, 3H, OCH3), 3.48 (s, 6H, OCH3), 3.50 (s, 6H, OCH3), 3.52 (s, 3H, OCH3), 3.66 (s, 18H, OCH3), 3.10–4.36 (35H, H-2, H-3, H-4, H-5, H-6B,C,E,F, H-6aA or D), 4.51 (dd, 3JH-6b,H-5 = 3.2 Hz, 2JH-6b,H-6a = 10.1 Hz, 1H, H-6bA or D), 5.00 (d, 3JH-1,H-2 = 3.2 Hz, 1H, H-1), 5.02 (d, 3JH-1,H-2 = 3.2 Hz, 1H, H-1), 5.08 (t, 4J = 1.7 Hz, 2H, OCH2Ph), 5.05–5.11 (4d, 3JH-1,H-2 = 3.2 Hz, 4H, H-1), 6.69 (s, 1H, OH ), 6.79–7.02 (8H, catecholate H), 7.21–7.44 (5H, arom. benzyl H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.56, 57.86 〚× 4〛, 58.12, 58.74, 58.81, 58.94, 59.23 (CH3O-2, CH3O-6), 61.56, 61.79 〚× 5〛 (CH3O-3), 68.20 (OCH2Ph), 70.80, 70.85, 70.94 〚× 3〛, 71.23〚× 3〛, 71.35, 71.46, 71.50, 71.55 (C-5, C-6), 81.26 〚× 4〛, 81.39 〚× 2〛, 82.02 〚× 5〛, 82.15 〚× 2〛, 82.24 〚× 2〛, 82.38, 82.70, 82.97 (C-2, C-3, C-4), 99.59, 99.78 〚× 2〛, 99.90, 100.11, 100.37 (C-1), 114.60, 115.06, 115.25, 115.78, 120.04, 121.32 〚× 2〛, 122.76 (catecholate CH), 126.76, 127.51, 128.37 (benzyl CH), 137.54, 146.56, 146.95 〚× 2〛, 148.89 (Cipso). Anal. calcd for C71H106O32 (1471.59): C 57.95, H 7.26; found: C 57.71, H 7.30.
Fraction 3. 6A,6D-di-O-(1,2-phenylene)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin (4a) (0.183 g, 17%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.15; Mp 127–130 °C; 1H NMR (500 MHz, CDCl3, 25 °C, assignment by ROESY and COSY; signals with unassigned multiplicity are overlapping with other signals): δ 2.90 (d, 2JH-6a,H-6b = 11.4 Hz, 2H, H-6aB,E), 3.12 (dd, 2H, 3JH-2,H-1 = 3.6 Hz, H-2B,E), 3.15 (dd, 2H, H-2C,F), 3.19 (dd, 2H, H-2A,D), 3.20 (s, 6H, OCH3), 3.30 (t, 2H, H-4A,D), 3.30 (s, 6H, OCH3), 3.46 (s, 6H, OCH3), 3.47 (s, 6H, OCH3), 3.51 (t, 2H, H-3C,F), 3.55 (s, 6H, OCH3), 3.56 (t, 2H, H-3A,D), 3.60 (dd, 2H, H-4B,E), 3.60 (dd, 2H, H-5C,F), 3.60 (dd, 2H, H-5B,E), 3.60 (s, 6H, OCH3), 3.62 (2H, H-6aC,F), 3.67 (2H, H-3B,E), 3.68 (s, 6H, OCH3), 3.74 (2H, H-6bC,F), 3.75 (s, 6H, OCH3), 3.78 (2H, H-4C,F), 3.89 (dd, 2H, 2JH-6b,H-6a = 11.4 Hz, 3JH-6b,H-5 = 2.3 Hz, H-6bB,E), 4.20 (2H, H-6bA,D), 4.24 (d2H, H-6aA,D), 4.33 (dd, 2H, 3JH-5,H-6a = 10.4 Hz, 3JH-5,H-4 = 4.5 Hz, H-5A,D), 4.96 (d, 3JH-1,H-2 = 3.2 Hz, 4H, H-1A,D), 4.96 (d, 3JH-1,H-2 = 3.2 Hz, 4H, H-1C,F), 5.23 (d, 3JH-1,H-2 = 3.6 Hz, 2H, H-1B,E), 6.83–6.94 (AA’BB’ system, 4H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.58 〚× 2〛, 58.93 〚× 2〛, 59.67 (CH3O-2, CH3O-6), 61.00, 61.68, 61.94 (CH3O-3), 69.39, 70.45, 71.39 (C-6), 71.03, 71.59, 72.37 (C-5), 79.56, 80.56, 80.92, 81.47 〚× 2〛, 81.68, 82.44, 82.65 〚× 2〛 (C-2, C-3, C-4), 97.16, 98.46, 99.59 (C-1), 112.87, 120.35 (arom. CH), 148.59 (Cipso). Anal. calcd for C58H94O30·C7H16 (1271.35 + 100.20): C 56.92, H 8.08; found: C 56.78, H 7.58.
Alkylation of 2-benzyloxyphenol (0.740 g, 3.69 mmol, 0.85 ml) with dimesylate 1b (1.000 g, 0.739 mmol) according to the previously described procedure afforded two major compounds as well as small amounts of debenzylation products, which were not separated.
Fraction 1. 6A,6C-di-O-(2-benzyloxyphenyl)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin (2b) (0.650 g, 57%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.36; Mp 111–113 °C; 1H NMR (300 MHz, CDCl3): δ = 3.15 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 3.32 (s, 3H, OCH3), 3.37 (s, 9H, OCH3), 3.47 (s, 3H, OCH3), 3.49 (s, 6H, OCH3), 3.51 (s, 3H, OCH3), 3.63 (s, 3H, OCH3), 3.64 (s, 6H, OCH3), 3.66 (s, 9H, OCH3), 3.08–4.54 (36H, H-2, H-3, H-4, H-5, H-6), 5.01–5.10 (10H, H-1, OCH2Ph), 6.82–6.96 (8H, catecholate H), 7.26–7.44 (10H, arom. benzyl H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.40 〚× 2〛, 57.76 〚× 2〛, 57.82 〚× 2〛, 58.81 〚× 2〛, 58.94 〚× 2〛 (CH3O-2, CH3O-6), 61.59 〚× 2〛, 61.66 〚× 4〛 (CH3O-3), 68.41 〚× 2〛 (OCH2Ph), 70.67 〚× 2〛 (C-6A,C), 70.84 〚× 2〛,71.13 〚×4〛 (C-5), 71.20 〚× 2〛, 71.33 〚× 2〛 (C-6B,D,E,F), 81.16 〚× 4〛, 81.95 〚× 2〛, 82.08 〚× 4〛, 82.24 〚× 4〛, 82.34 〚× 2〛, 82.42 〚× 2〛 (C-2, C-3, C-4), 99.75 〚× 2〛 (C-1A,C), 100.08 〚× 4〛, (C-1B,D,E,F), 114.60 〚× 2〛, 114.96 〚× 2〛, 121.22 〚× 2〛, 121.35 〚× 2〛 (catecholate CH), 126.73 〚× 2〛, 127.48 〚× 2〛, 128.33 〚× 2〛 (benzyl CH), 137.41 〚× 2〛, 148.83 〚× 2〛, 149.25 〚× 2〛 (Cipso) (the 13C NMR spectrum shows an apparent C2 symmetry). Anal. calcd for C78H112O32 (1561.71): C 59.99, H 7.23; found: C 59.79, H 7.28.
Fraction 2. 6A,6C-di-O-(1,2-phenylene)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin (4b) (0.183 g, 18%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.25; Mp 124–126 °C; 1H NMR (300 MHz, CDCl3): δ = 3.03 (s, 3H, OCH3), 3.20 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 3.38 (s, 3H, OCH3), 3.48 (s, 6H, OCH3), 3.49 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.50 (s, 3H, OCH3), 3.56 (s, 3H, OCH3), 3.60 (s, 6H, OCH3), 3.64 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 3.70 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.11–4.41 (36H, H-2, H-3, H-4, H-5, H-6), 4.97 (d, 3JH-1,H-2 = 3.7 Hz, 1H, H-1), 5.00 (d, 3JH-1,H-2 = 3.3 Hz, 1H, H-1), 5.05 (d, 3JH-1,H-2 = 2.7 Hz, 2H, H-1), 5.06 (d, 3JH-1,H-2 = 2.5 Hz, 1H, H-1), 5.08 (d, 3JH-1,H-2 = 3.9 Hz, 1H, H-1) 5.33 (d, 3JH-1,H-2 = 3.7 Hz, 1H, H-1), 6.81–7.00 (4H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.39, 57.45, 57.62, 58.29, 58.46 〚× 2〛, 58.58, 58.83 〚× 2〛, 59.54 (CH3O-2, CH3O-6) 60.75, 61.24, 61.39 〚× 2〛, 61.63, 61.78 (CH3O-3), 68.01, 68.81, 71.24, 71.62, 73.09 〚× 2〛 (C-6), 70.68 〚× 3〛, 70.82, 70.99 〚× 2〛 (C-5), 79.03, 79.89, 80.37, 80.82, 81.05 〚× 3〛, 81.36 〚× 3〛, 81.62, 81.75, 81.87, 82.03, 82.25 〚× 2〛, 82.51, 82.75 (C-2, C-3, C-4), 94.71, 97.58, 98.82, 98.87, 99.52, 99.99 (C-1), 112.83, 118.98, 120.46, 123.03 (arom. CH), 148.00, 151.13 (Cipso). Anal.calcd for C58H94O30 (1271.35): C 54.80, H 7.45; found: C 55.01, H 7.55.
4.2.4 Syntheses of 6A,6D-bis-O-(2-hydroxyphenyl)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D, 3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin (6a) and 6A,6C-bis-O-(2-hy droxyphenyl)-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin (6b)
Pd/C (10%, 0.100 g) was added to a solution of 2a (0.869 g) in EtOH (100 ml). The mixture was stirred for 48 h under H2 (1 atm.) before being filtered over celite. Evaporation of the filtrate in vacuo yielded pure 6a (0.717 g, 94%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.24; Mp 136–139 °C; 1H NMR (200 MHz, CDCl3): δ = 3.23 (s, 6H, OCH3), 3.30 (s, 6H, OCH3), 3.49 (s, 6H, OCH3), 3.51 (s, 6H, OCH3), 3.52 (s, 6H, OCH3), 3.65 (s, 12H, OCH3), 3.67 (s, 6H, OCH3), 3.13–4.45 (36H, H-2, H-3, H-4, H-5, H-6), 5.02 (d, 3JH-1,H-2 = 3.2 Hz, 2H, H-1), 5.08 (d, 3JH-1,H-2 = 3.7 Hz, 2H, H-1), 5.10 (d, 3JH-1,H-2 = 3.7 Hz, 2H, H-1), 6.60 (s, 2H, OH), 6.73–7.02 (8H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.92 〚×2〛, 58.26, 58.87, 59.03 (CH3O-2, CH3O-6), 61.77, 61.90 〚×2〛 (CH3O-3), 70.93, 71.40 〚×2〛 (C-6), 71.59 〚×3〛 (C-5), 81.23, 81.33, 81.40, 81.92 〚×2〛, 82.24 〚×2〛, 82.44, 82.67 (C-2, C-3, C-4), 99.77 〚×2〛, 100.48 (C-1), 115.53, 115.84, 120.09, 122.88 (arom. CH), 146.66, 147.05 (Cipso). Anal. calcd for C64H100O32 (1381.46): C 55.64, H 7.30; found: C 55.63, H 7.05.
Hydrogenolysis of 2b (0.650 g, 0.42 mmol) was carried out according to the above procedure to afford 6b as a colourless solid (0.540 g, 94%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.28; Mp 102–104 °C; 1H NMR (300 MHz, CDCl3): δ = 3.14 (s, 3H, OCH3), 3.26 (s, 3H, OCH3), 3.31 (s, 3H, OCH3), 3.33 (s, 3H, OCH3), 3.47 (s, 3H, OCH3), 3.48 (s, 6H, OCH3), 3.50 (s, 6H, OCH3), 3.52 (s, 3H, OCH3), 3.63 (s, 6H, OCH3), 3.64 (s, 6H, OCH3), 3.66 (s, 6H, OCH3), 3.19–4.46 (36H, H-2, H-3, H-4, H-5, H-6), 5.01 (d, 3JH-1,H-2 = 3.8 Hz, 1H, H-1), 5.02 (d, 3JH-1,H-2 = 3.8 Hz, 1H, H-1), 5.04 (d, 3JH-1,H-2 = 3.0 Hz, 1H, H-1), 5.08 (3 overlapping d, 3H, H-1), 6.61 (br s, 1H, OH), 6.67 (br s, 1H, OH), 6.74–7.02 (8H, arom. H); C{1H} NMR (50 MHz, CDCl3): δ = 57.75 〚× 3〛, 57.84, 58.01 〚× 2〛, 58.75, 58.87 〚× 2〛, 59.03 (CH3O-2, CH3O-6), 61.65 〚× 5〛, 61.83 (CH3O-3), 70.56, 70.83 〚× 4〛, 71.30 (C-6), 71.07, 71.35 〚× 4〛, 71.73 (C-5), 81.20 〚× 4〛, 81.32, 81.49, 81.75, 81.89, 82.05 〚× 2〛, 82.11 〚× 3〛, 82.22, 82.49 〚× 2〛, 82.66, 83.10 (C-2, C-3, C-4), 99.38, 99.72, 99.82 〚× 2〛, 100.21 〚× 2〛 (C-1), 115.16, 115.62 〚× 2〛, 115.89, 119.85, 120.02, 122.57, 122.80 (arom. CH), 146.47, 146.61, 146.86 〚× 2〛 (Cipso). Anal. calcd for C64H100O32 (1381.46): C 55.64, H 7.30; found: C 55.41, H 7.24.
4.2.5 Syntheses of 6A,6D-bis-O-{2-〚(diphenoxyphosphino)oxy〛phenyl}-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin (L1) and 6A,6C-bis-O-{2-〚(diphenoxyphosphino)oxy〛phenyl}-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin (L2)
Chlorodiphenylphosphite (0.220 g, 0.89 mmol, 0.18 ml) was added to a stirred solution of 6a (0.500 g, 0.36 mmol) in CH2Cl2 (25 ml) at –40 °C. The reaction mixture was stirred for 15 min at –40 °C whereupon triethylamine (0.18 ml, 0.90 mmol, 1 equiv) was added. After 1 h, addition of pentane (200 ml) caused the triethylammonium salt to precipitate. The latter was filtered off and the filtrate was evaporated to dryness under vacuum. The residue was washed with boiling hexane (10 ml). The hot suspension was then cooled down to 0 °C and allowed to settle whereupon the hexane phase was discarded. The colourless residue was found to be pure L1 (0.630 g, 96%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.31; Mp 83–86 °C; 1H NMR (200 MHz, CDCl3): δ = 3.34 (s, 6H, OCH3), 3.35 (s, 6H, OCH3), 3.38 (s, 6H, OCH3), 3.39 (s, 6H, OCH3), 3.48 (s, 6H, OCH3), 3.62 (s, 6H, OCH3), 3.65 (s, 12H, OCH3), 3.01–4.19 (34H, H-2, H-3, H-4, H-5, H-6a, H-6bB,C,E,F), 4.90 (d, 2JH-6a,H-6b = 11.6 Hz, 2H, H-6bA,D), 4.93 (d, 3JH-1,H-2 = 3.0 Hz, 2H, H-1), 5.02 (d, 3JH-1,H-2 = 3.2 Hz, 2H, H-1), 5.10 (d, 3JH-1,H-2 = 2.5 Hz, 2H, H-1), 6.84–7.40 (28H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.27, 57.89 〚× 2〛 (CH3O-2), 58.94, 59.13 (CH3O-6), 61.72 〚× 3〛 (CH3O-3), 67.95 (C-6A,D), 70.84, 71.26, 71.39 (C-5), 71.62, 72.15 (C-6B,C,E,F), 81.10, 81.23 〚× 2〛, 81.65, 81.82, 81.92, 82.28, 82.47, 82.97 (C-2, C-3, C-4), 99.42 (C-1A,D), 100.24, 100.47 (C-1B,C,E,F), 113.98 (s, Cpara), 120.80 (d, 3JC,P = 6.6 Hz, Cortho), 121.37 〚× 2〛 (d, 4JC,P = 4.9 Hz, Cmeta), 122.45 (d, 4JC,P = 4.9 Hz, Cmeta), 124.02 〚× 2〛 (d, 4JC,P = 4.9 Hz, Cpara), 124.63 (s, Cmeta), 129.47 〚× 2〛 (d, 3JC,P = 8.2 Hz, Cortho), 141.22 (d, 2JC,P = 3.3 Hz, Cipso), 150.23 (d, 2JC,P = 3.3 Hz, Cipso), 151.48 (s, Cipso), 151.81 (d, 2JC,P = 3.3 Hz, Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = 127.5 (s). Anal. calcd for C88H118O36P2 (1813.81): C 58.27, H 6.56; found: C 58.32, H 6.36; MS (FAB): m/z (%): 1813.6 (50) 〚M+H〛+.
Ligand L2 was prepared in 99% yield (0.700 g) as a colourless solid according to the above procedure from chlorodiphenylphosphite (0.247 g, 0.98 mmol, 0.20 ml) and 6b (0.540 g, 0.39 mmol). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.38; Mp 61–63 °C; 1H NMR (300 MHz, CDCl3): δ = 3.29 (s, 3H, OCH3), 3.32 (s, 3H, OCH3), 3.33 (s, 3H, OCH3), 3.34 (s, 3H, OCH3), 3.34 (s, 6H, OCH3) 3.36 (s, 3H, OCH3), 3.41 (s, 3H, OCH3), 3.47 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.59 (s, 3H, OCH3), 3.61 (s, 3H, OCH3), 3.62 (s, 3H, OCH3), 3.63 (s, 3H, OCH3), 3.64 (s, 6H, OCH3), 2.98–4.18 (34H, H-2, H-3, H-4, H-5, H-6a, H-6bB,D,E,F), 4.75 (dd, 2JH-6a,H-6b = 10.5 Hz, 2H, H-6bA or C), 4.80 (dd, 2JH-6a,H-6b = 10.7 Hz, 2H, H-6bC or A), 4.85 (d, 3JH-1,H-2 = 3.4 Hz, 1H, H-1), 4.91 (d, 3JH-1,H-2 = 3.1 Hz, 1H, H-1), 5.01 (d, 3JH-1,H-2 = 3.1 Hz, 1H, H-1), 5.05 (d, 3JH-1,H-2 = 3.4 Hz, 1H, H-1), 5.07 (d, 3JH-1,H-2 = 2.8 Hz, 1H, H-1), 5.08 (d, 3JH-1,H-2 = 3.0 Hz, 1H, H-1), 6.84–7.35 (28H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.27 〚× 2〛, 57.82 〚× 4〛 (CH3O-2), 58.81, 58.91, 59.00 〚× 2〛 (CH3O-6), 61.66 〚× 6〛 (CH3O-3), 67.85 〚× 2〛 (C-6A,C), 70.51 〚× 2〛, 70.67 〚× 2〛, 71.30 〚× 2〛 (C-5), 71.50 〚× 2〛, 71.92 〚× 2〛 (C-6B,D,E,F), 81.16 〚× 8〛, 81.62 〚× 2〛, 81.85 〚× 2〛, 82.18 〚× 2〛, 82.34, 82.57 〚× 2〛, 82.83 (C-2, C-3, C-4), 99.42 (C-1A,C), 100.11 〚× 2〛, 100.27 〚× 2〛 (C-1B,C,E,F), 113.81 〚× 2〛 (s, Cpara), 120.74 〚×2〛 (d, 3JC,P = 8.2 Hz, Cortho), 121.26 〚× 4〛 (d, 4JC,P = 6.6 Hz, Cmeta), 122.38 〚× 2〛 (d, 4JC,P = 4.9 Hz, Cmeta), 123.98 〚× 4〛 (d, 4JC,P = 3.3 Hz, Cpara), 124.56 〚× 2〛 (s, Cmeta), 129.42 〚× 4〛 (d, 3JC,P = 9.9 Hz, Cortho), 141.19 〚× 2〛 (d, 2JC,P = 3.3 Hz, Cipso), 150.13 〚× 2〛 (s, Cipso), 151.44 〚× 2〛 (s, Cipso), 151.74 〚× 2〛 (d, 2JC,P = 3.3 Hz, Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = 127.5 〚× 2〛 (s). Anal. calcd for C88H118O36P2 (1813.81): C 58.27, H 6.56; found: C 58.32, H 6.36.
4.2.6 Syntheses of P,P’-(6A,6D-di-O-{2-〚(diphenoxyphosphino)oxy〛phenyl}-2A,2B, 2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin)-bis〚chloro(o-dimethylaminobenzyl)palladium(II)〛 (9a) and P,P’-(6A,6C-di-O-{2-〚(diphenoxyphosphino)oxy〛phenyl}-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin)-bis〚chloro(o-dimethylaminobenzylpalladium(II)〛 (9b)
〚(o-C6H4CH2NMe2)PdCl〛2 (0.030 g, 0.055 mmol) was added to a solution of L1 (0.100 g, 0.055 mmol) in CH2Cl2 (5 ml). After 20 min, the product was precipitated by addition of pentane (250 ml) and then collected on Celite. The precipitate was dissolved in CH2Cl2 (15 ml), and upon filtration, the organic solution was evaporated to dryness in vacuo to afford pure 9a as a yellow powder (0.073 g, 56%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.40; Mp 123–125 °C; 1H NMR (200 MHz, CDCl3): δ = 2.61 (d, 6 H, 4JH,P = 3.9 Hz, NCH3), 2.63 (d, 6H, 4JH,P = 4.2 Hz, NCH3), 3.30 (s, 6H, OCH3), 3.33 (s, 12H, OCH3), 3.35 (s, 6H, OCH3), 3.47 (s, 6H, OCH3), 3.60 (s, 6H, OCH3), 3.63 (s, 12H, OCH3), 2.84–4.15 (38H, H-2, H-3, H-4, H-5, H-6a, H-6bB,C,E,F, CH2Ph), 4.86 (d, 2JH-6a,H-6b = 9.3 Hz, 2H, H-6bA,D), 5.01 (2d, 3JH-1,H-2 = 2.9 Hz, 4H, H-1B,C,E,F), 5.13 (d, 3JH-1,H-2 = 3.2 Hz, 2H, H-1A,D), 6.83–7.72 (36H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 50.00 (d, 3JC,P = 3.3 Hz, NCH3), 50.15 (d, 3JC,P = 3.3 Hz, NCH3), 57.40, 57.86 〚× 2〛 (CH3O-2), 58.91, 59.17 (CH3O-6), 61.53, 61.66, 61.72 (CH3O-3), 67.69 (C-6A,D), 70.90, 71.17, 71.39 (C-5), 71.59, 72.21 (C-6B,C,E,F), 72.68 (CH2Ph), 81.23 〚× 3〛, 81.65, 81.75, 81.79, 82.21, 82.28, 82.93 (C-2, C-3, C-4), 99.39 (C-1A,D), 100.14, 100.44 (C-1B,C,E,F), 114.04 (s, Cpara), 120.67 (s, DMBA), 121.21 (d, 4JC,P = 4.9 Hz, Cmeta), 121.95 (d, 4JC,P = 6.6 Hz, DMBA), 122.41 (s Cortho), 124.57 (s, Cmeta), 124.80 (s, Cpara), 125.23 (s, Cmeta), 125.92 (d, 4JC,P = 6.6 Hz, DMBA), 129.29 (d, 3JC,P = 3.3 Hz, Cortho), 136.92 (d, 3JC,P = 11.5 Hz, DMBA), 140.81 (d, 2JC,P = 4.9 Hz, Cipso), 147.62 (d, 2JC,P = 3.3 Hz, Cipso), 149.83 〚× 2〛 (d, 2JC,P = 4.9 Hz, Cipso), 151.06 (d, 2JC,P = 3.3 Hz, Cipso), 151.22 (s, Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = 104.5 (s). Anal. calcd for C106H142Cl2N2O36P2Pd2 (2365.96): C 53.81, H 6.05; found: C 54.07, H 6.07.
Complex 9b was prepared in 75% yield (0.100 g) according to the above procedure from L2 (0.103 g, 0.057 mmol) and 〚(o-C6H4CH2NMe2)PdCl〛2 (0.031 g, 0.057 mmol). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.30; Mp 97–99 °C; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 2.60 (d, 6 H, 4JH,P = 3.9 Hz, NCH3), 2.62 (d, 6H, 4JH,P = 3.7 Hz, NCH3), 3.21 (s, 3H, OCH3), 3.30 (s, 3H, OCH3), 3.31 (s, 6H, OCH3), 3.32 (s, 3H, OCH3), 3.34 (s, 3H, OCH3), 3.35 (s, 3H, OCH3), 3.40 (s, 3H, OCH3), 3.46 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.59 (s, 3H, OCH3), 3.60 (s, 3H, OCH3), 3.62 (s, 6H, OCH3), 3.64 (s, 3H, OCH3), 3.65 (s, 3H, OCH3), 2.94–4.17 (38H, H-2, H-3, H-4, H-5, H-6a, H-6bB,D,E,F, CH2Ph), 4.73 (dd, 2J H-6a,H-6b = 10.6 Hz, 3J H-6a,H-5 = 3.0 Hz, 1H, H-6bA or C), 4.79 (dd, 2JH-6a,H-6b = 10.6 Hz, 3J H-6a,H-5 = 2.8 Hz, 1H, H-6bC or A), 4.94 (d, 3JH-1,H-2 = 3.4 Hz, 1H, H-1), 4.99 (2d, 3JH-1,H-2 = 2.6 Hz, 1H, H-1), 5.00 (2d, 3JH-1,H-2 = 2.9 Hz, 1H, H-1), 5.05 (d, 3JH-1,H-2 = 3.4 Hz, 1H, H-1), 5.11 (2d, 3JH-1,H-2 = 3.1 Hz, 2H, H-1), 6.85–6.91 (8H, arom. H), 6.98–7.10 (8H, arom. H), 7.20–7.33 (18H, arom. H), 7.63–7.72 (2H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 49.98 (d, 3JC,P = 3.3 Hz, NCH3), 50.14 (d, 3JC,P = 4.9 Hz, NCH3), 57.40 〚× 2〛, 57.79 〚× 4〛 (CH3O-2), 58.81, 58.94, 59.00, 59.07 (CH3O-6), 61.50 〚× 2〛, 61.65, 61.69 〚× 2〛, 61.76 (CH3O-3), 67.59 (C-6A,C), 70.61, 70.71, 71.26 (C-5), 71.39, 71.98 (C-6B,D,E,F), 72.64, 72.74 (CH2Ph), 81.20 〚× 2〛, 81.69 〚× 2〛, 81.85, 82.18, 82.28, 82.51, 82.93 (C-2, C-3, C-4), 99.45 (C-1A,C), 100.08, 100.31 (C-1B,D,E,F), 113.88 〚× 2〛 (s, Cpara), 120.63 〚× 2〛 (s, DMBA), 121.17 〚× 4〛 (d, 4JC,P = 4.9 Hz, Cmeta), 122.03 〚× 2〛 (d, 4JC,P = 8.2 Hz, DMBA), 122.37 〚× 2〛 (s Cortho), 124.50 〚× 2〛 (s, Cmeta), 124.80 〚× 4〛 (d, 5JC,P = 3.3 Hz, Cpara), 125.19 〚× 2〛 (s, Cmeta), 125.86 〚× 2〛 (d, 4JC,P = 8.2 Hz, DMBA), 129.22 〚× 4〛 (d, 3JC,P = 3.3 Hz, Cortho), 136.88 〚× 2〛 (d, 3JC,P = 11.5 Hz, DMBA), 140.77 〚× 2〛 (d, 2JC,P = 4.9 Hz, Cipso), 147.55 〚× 2〛 (d, 2JC,P = 3.3 Hz, Cipso), 149.72 〚× 4〛 (d, 2JC,P = 4.9 Hz, Cipso), 151.02 〚× 2〛 (d, 2JC,P = 3.3 Hz, Cipso), 151.18 〚× 2〛 (s, Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = 104.6 〚×2〛 (s). Anal. calcd for C106H142Cl2N2O36P2Pd2 (2365.96): C 53.81, H 6.05; found: C 53.78, H 6.16.
4.2.7 Syntheses of P,P’-(6A,6D-bis-O-{2-〚(diphenoxyphosphino)oxy〛phenyl}-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin) silver(I) tetrafluoroborate (10a) and P,P’-(6A,6C-bis-O-{2-〚(diphenoxyphosphino)oxy〛phenyl}-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E,6F-hexadeca-O-methyl-α-cyclodextrin)silver(I) tetrafluoroborate (10b)
A solution of AgBF4 (0.011 g, 0.055 mmol) in THF (20 ml) was added to a solution of L1 (0.100 g, 0.055 mmol) in CH2Cl2 (200 ml) under vigorous stirring. After 20 min, the reaction mixture was concentrated to 5 ml and pentane (250 ml) was added to precipitate the product, which was collected on celite. The precipitate was dissolved in CH2Cl2 (15 ml), and upon filtration, the organic solution was evaporated to dryness in vacuo to afford pure 10a as a colourless powder (0.060 g, 55%). 1H NMR (200 MHz, CDCl3): δ = 3.16 (s, 6H, OCH3), 3.37 (s, 6H, OCH3), 3.41 (s, 6H, OCH3), 3.42 (s, 6H, OCH3), 3.44 (s, 6H, OCH3), 3.59 (s, 6H, OCH3), 3.61 (s, 12H, OCH3), 2.86–4.67 (36H, H-2, H-3, H-4, H-5, H-6), 4.92 (d, 3JH-1,H-2 = 2.9 Hz, 2H, H-1), 5.02 (d, 3JH-1,H-2 = 3.0 Hz, 2H, H-1), 5.07 (d, 3JH-1,H-2 = 3.2 Hz, 2H, H-1), 6.90–7.46 (28H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.59, 57.79, 57.99 (CH3O-2), 60.28 〚× 2〛 (CH3O-6), 61.53, 61.63, 61.65 (CH3O-3), 68.71 (C-6A,D), 69.69, 70.67, 71.26 (C-5), 71.59, 71.85 (C-6B,C,E,F), 80.77, 81.29 〚× 3〛, 82.02 〚× 5〛 (C-2, C-3, C-4), 99.95 〚× 2〛 (C-1B,C,E,F), 101.09 (C-1A,D), 115.58 (s, Cpara), 121.06 (d, 3JC,P = 6.6 Hz, Cortho), 121.26 〚× 2〛 (d, 4JC,P = 6.6 Hz, Cmeta), 122.60 (d, 4JC,P = 3.3 Hz, Cmeta), 126.17 〚× 2〛 (s, Cpara), 127.32 (s, Cmeta), 130.40 〚× 2〛 (d, 4JC,P = 6.6 Hz, Cortho), 138.40 (s, Cipso), 149.74 〚× 2〛 (d, 2JC,P = 3.3 Hz, Cipso), 150.04 (d, 2JC,P = 3.3 Hz, Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = 116.7 〚2d, 107JAg,P = 1083 Hz, 109JAg,P = 1231 Hz〛. Anal. calcd for C88H118AgBF4O36P2·1.5 CHCl3 (2008.48 + 179.07): C 49.14, H 5.51; found: C 49.05, H 5.38; MS (FAB): m/z (%): 1937.3 (20) 〚M–BF4+O〛+, 1921.3 (100) 〚M–BF4〛+.
Complex 10b was prepared in 81% yield (0.090 g) according to the above procedure from L2 (0.11 g, 0.056 mmol) and AgBF4 (0.011 g, 0.056 mmol). 1H NMR (300 MHz, CDCl3): δ = 3.40 (s, 6H, OCH3), 3.43 (s, 6H, OCH3), 3.47 (s, 6H, OCH3), 3.56 (s, 6H, OCH3), 3.60 (s, 6H, OCH3), 3.62 (s, 6H, OCH3), 3.63 (s, 6H, OCH3), 3.64 (s, 6H, OCH3), 2.86–4.25 (36H, H-2, H-3, H-4, H-5, H-6), 4.96 (br d, 1H, H-1), 4.99 (2d, 3JH-1,H-2 = 3.2 Hz, 2H, H-1), 5.04 (br d, 1H, H-1), 5.11 (br d, 1H, H-1), 5.20 (br d, 1H, H-1), 6.60–7.60 (28H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = 57.43 〚× 2〛, 57.53 〚× 2〛, 57.76 〚× 2〛 (CH3O-2), 58.91 〚× 2〛, 59.04 〚× 2〛 (CH3O-6), 61.66 〚× 2〛, 61.72 〚× 2〛, 61.79 〚× 2〛 (CH3O-3), 70.21 〚× 2〛, 70.90 〚× 4〛, 71.17 〚× 2〛, 71.50 〚× 2〛 (C-5, C-6), 80.97 〚× 2〛, 81.23 〚× 6〛, 82.02 〚× 2〛, 82.10 〚× 2〛, 82.28 〚× 6〛 (C-2, C-3, C-4), 100.11 〚×6〛 (C-1), 115.35–129.94 (arom. CH), 140.00–150.26 (Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = 108.3 and 114.5 (ABX, 107JAg,P = 1095 Hz, 109JAg,P = 1203 Hz and 107JAg,P’ = 977 Hz, 109JAg,P’ = 1088 Hz, 2JP,P’ = 256 Hz, P and P’). Anal. calcd for C88H118AgBF4O36P2·0.75 (CH3)2CO (2008.48 + 43.56): C 52.83, H 6.02; found: C 53.10, H 6.09; MS (FAB): m/z (%): 1937.3 (20) 〚M–BF4+O〛+, 1921.3 (100) 〚M–BF4〛+.
4.2.8 Syntheses of cis-P,P’-(6A,6D-bis-O-{2-〚(diphenoxyphosphino)oxy〛pheny}-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6C,6E,6F-hexadeca-O-methyl-α-cyclodextrin)-〚(norbornadiene)rhodium(I)〛 hexafluorophosphate (11a) and cis-P,P’-(6A,6C-bis-O-{2-〚(diphenoxyphosphino)oxy〛pheny}-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6E, 6F-hexadeca-O-methyl-α-cyclodextrin)-〚(norbornadiene)rhodium(I)〛 tetrafluoroborate (11b)
Powdered TlPF6 (0.022 g, 0.063 mmol) was added to a solution of 〚RhCl(NBD)〛2 (0.013 g, 0.028 mmol) in THF/CH2Cl2 (50:50, v/v, 50 ml). After stirring the suspension vigorously for 15 min, the precipitate was collected on celite and the filtrate was directly added to a solution of L1 (0.106 g, 0.058 mmol) in CH2Cl2 (200 ml) under vigorous stirring. The reaction mixture was then stirred at room temperature for 20 min before being concentrated to 5 ml. Addition of pentane (250 ml) caused the product to precipitate. The solid was then filtered over celite before being dissolved in CH2Cl2 (15 ml). Upon filtration, the organic solution was evaporated to dryness in vacuo to afford pure 11a as an orange powder (0.110 g, 90%). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.10; Mp 118 °C dec; 1H NMR (500 MHz, CDCl3, 54 °C): δ = 3.21 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 3.25 (s, 3H, O-CH3), 3.26 (s, 3H, OCH3), 3.35 (s, 3H, OCH3), 3.37 (s, 6H, OCH3), 3.40 (s, 3H, OCH3), 3.48 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.60 (s, 3H, OCH3), 3.61 (s, 3H, OCH3), 3.64 (s, 6H, OCH3), 3.66 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 2.95–5.04 (50 H, H-1, H-2, H-3, H-4, H-5, H-6, HC=CH and CH of NBD), 6.75–7.50 (28H, arom. H); 13C{1H} NMR (50 MHz, CDCl3): δ = (all signals are broad) 47.24 (CH2 of NBD), 57.66 (CH3O-2), 58.71, 59.10 (CH3O-6), 61.72 (CH3O-3), 69.82 (CH of NBD), 71.13 (C-5, C-6), 81.29 (HC=CH of NBD), 82.27 (C-2, C-3, C-4), 100.18 (C-1), 115.32, 119.29, 120.53, 126.10, 127.25, 129.68, 130.30 (arom. CH), 138.63, 150.53 (Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = –145 (hept. PF6), 110.5 (d, JRh,P = 264 Hz). Anal. calcd for C95H126F6O36P3Rh·CHCl3 (2153.86 + 119.38): C 50.72, H 5.63; found: C 50.72, H 5.73; MS (FAB): m/z (%): 2023.6 (37) 〚M–PF6+O〛, 2007.7 (49) 〚M–PF6〛+, 1915.5 (58) 〚M–PF6-NBD〛+.
Complex 11b was prepared in 83% yield (0.120 g) according to the above procedure from L2 (0.125 g, 0.069 mmol) and the in situ prepared 〚Rh(THF)2(NBD)〛BF4 obtained from treatment of 〚RhCl(NBD)〛2 (0.016 g, 0.035 mmol) with AgBF4 (0.013 g, 0.067 mmol). Rf (CH2Cl2/MeOH, 94:6, v/v) = 0.30; Mp 155° dec. All 1H signals are broad. 13C{1H} NMR (50 MHz, CDCl3): all signals are broad, δ = 57.46 (CH3O-2), 58.81 (CH3O-6), 61.63 (CH3O-3), 70.71 (C-5, C-6), 81.36, 82.31 (C-2, C-3, C-4), 100.18 (C-1), 115.35, 120.47, 126.07, 129.28, 129.55, 130.27 (arom. CH), 138.50, 150.43 (Cipso); 31P{1H} NMR (121.5 MHz, CDCl3): δ = 110.5 (d, JRh,P = 259 Hz). Anal. calcd for C95H126BF4O36Rh (2096.64): C 54.42, H 6.06; found: C 54.21, H 6.10; MS (FAB): m/z (%): 2024.4 (10) 〚M–BF4+O〛+, 2008.4 (18) 〚M–BF4〛+, 1915.3 (40) 〚M–BF4-NBD〛+.
4.3 X-ray crystallography
Crystals of compound 4a suitable for diffraction study were obtained by slow diffusion of heptane into an EtOAc solution of the compound.
Crystal data. C58H94O30·C7H16, M = 1371.52, hexagonal, space group P6222, colourless crystals, a = 31.2588(2), c = 15.0373(1) Å, V = 12724.6(1) Å–3, Z = 6, Dx = 1.072 mg m–3, μ = 0.84 cm–1, F(000) = 4428. Data were collected on a Nonius KappaCCD (graphite-monochromated Mo Kα radiation 0.710 73 Å) at 100 K. 119 971 reflections collected, 4864 with I > 2 σ(I). The structure was solved with SHELLXD 〚29〛, which revealed the non-hydrogen atoms of the structure. After anisotropic refinement of the cyclodextrin moiety, the bridging aromatic ring appeared as having two well-defined orientations each of 50% occupancy. The presence of a disordered n-heptane chain in the cavity was also detected. The whole structure was refined with SHELLXL by the full-matrix least-squares techniques (use of F2; x, y, z, βij for the cyclodextrin O and C atoms, x, y, z, Biso for the other non-H atoms and x, y, z in riding mode for H atoms; 391 variables, 4864 observations with I > 2.0 σ(I), w = 1/〚σ2(Fo2) + (0.2 P)2〛 where P = 〚Fo2 + 2 Fc2〛/3). Final results R = 0.122, wR = 0.331 and Sw = 1.32.
Supplementary material
The supplementary material has been sent to the Cambridge Crystallographic Data centre, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary material No. 178633 and can be obtained by contacting the CCDC (quoting the article details and the corresponding SUP number).
Acknowledgements
É.E. thanks the ‘Ministère de la Culture, de l’Enseignement supérieur et de la Recherche’ of the Grand-Duchy of Luxembourg for a grant. We thank Johnson Matthey for a generous gift of rhodium trichloride and Wacker Chemie for a supply of α-cyclodextrin.